










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkEstudios Gerenciales
Print version ISSN 0123-5923
estud.gerenc. vol.31 no.134 Cali Jan./Mar. 2015
ARTÍCULO
La calidad desde el diseño: principios y oportunidades para la industria farmacéutica
Quality by design: Principles and opportunities for the pharmaceutical industry
A qualidade desde o projecto: princípios e oportunidades para a indústria farmacêutica
Oscar Fabián García Apontea*, Bibiana Margarita Vallejo Díazb, Claudia Elizabeth Mora Huertasc
a Estudiante, Maestría en Ingeniería Industrial, Universidad Nacional de Colombia, Bogotá D.C., Colombia
* Autor para correspondencia: Cra 30 No 45-00, Ed. 450, Universidad Nacional de Colombia, Bogotá, Colombia. Correo electrónico: ofgarciaa@unal.edu.co (O.F. García Aponte).
b Investigadora, Grupo de Investigación en Procesos de Transformación de Materiales, Departamento de Farmacia, Universidad Nacional de Colombia, Bogotá D.C., Colombia
c Investigadora, Grupo de Investigación en Desarrollo y Calidad de Productos Farmacéuticos y Cosméticos, Departamento de Farmacia, Universidad Nacional de Colombia, Bogotá D.C., Colombia
Recibido: el 14 de febrero de 2014
Aceptado: el 12 de septiembre de 2014
RESUMEN
La calidad desde el diseño ofrece ventajas sobre modelos previos de aseguramiento de la calidad, siendo útil en sectores donde la calidad signifique diferenciación y la flexibilización de los procesos estimule el mejoramiento continuo y la innovación. Como una contribución para facilitar su implementación en la industria farmacéutica en Colombia, en el presente artículo se analizan las circunstancias que la originaron. Igualmente, partiendo de un estudio de literatura especializada, se explica su importancia en el desarrollo de productos y se proponen algunos aspectos metodológicos para su puesta en práctica. La evidencia reportada sugiere que esta nueva visión de la calidad es una estrategia que genera confianza en clientes, industria y organismos reguladores, respecto a la calidad de los productos farmacéuticos.
Palabras clave: Calidad desde el diseño. Industria farmacéutica. Gestión de la calidad.
Códigos JEL L65.
ABSTRACT
Quality by design offers advantages over previous quality assurance models, being useful in areas where quality means the differentiation and implementation of flexible processes to stimulate continuous improvement and innovation. As a contribution to making quality by design easier to implement in the Colombian pharmaceutical industry, this paper discusses why this concept was originated. Likewise, based on a literature analysis, the noteworthiness of quality by design when products are being developed is discussed, and methodological strategies for its accomplishment are proposed. The reported evidence suggests that quality by design builds consumer, industrial and regulatory confidence on the quality of pharmaceutical products.
Keywords: Quality by design. Pharmaceutical industry. Quality management.
JEL classification L65.
RESUMO
A qualidade desde o projecto oferece vantagens sobre modelos anteriores de garantia de qualidade, sendo útil em sectores onde a qualidade significa diferenciação e a flexibilização dos processos estimula a melhora contínua e a inovação. Como uma contribuição para facilitar a sua implementação na indústria farmacêutica na Colômbia, analisam-se no presente artigo as circunstancias que a originaram. Igualmente, a partir de um estudo de literatura especializada, explica-se a sua importância no desenvolvimento de produtos e propõem-se alguns aspectos metodológicos para se pôr em prática. A evidência referida sugere que esta nova visão da qualidade é uma estratégia que gera confiança nos clientes, indústria e organismos reguladores, sobre a qualidade dos produtos farmacêuticos.
Palavras-chave: Qualidade desde o projecto. Indústria farmacêutica. Gestão da qualidade.
Classificações JEL L65.
1. Introducción
La consolidación de la gestión del conocimiento como un factor determinante en la competitividad de las industrias del mundo globalizado ha cuestionado sus paradigmas acerca de la implementación de los conceptos de aseguramiento de la calidad, desarrollo de producto y productividad, y su apropiación en una compañía ha constituido una característica diferenciadora y generadora de ventajas competitivas. Para el caso de la industria farmacéutica, la gestión del conocimiento ha significado la evolución desde la calidad por ensayo (Quality by Test [QbT]) a la calidad desde el diseño (Quality by Design [QbD]), que fue propuesta por primera vez en 1992 por Joseph Juran en su libro Juran on quality by design: the new steps for planning quality into goods and services (Juran, 1992).
Desde la óptica de la International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), QbD es un acercamiento sistemático al desarrollo farmacéutico, que parte de objetivos predefinidos, haciendo énfasis en el conocimiento del producto y en la comprensión y el control de los procesos. En este sentido, QbD se fundamenta en el empleo de ciencia verificable y en la toma de decisiones en función de la gestión del riesgo en calidad. De esta forma se fortalece el aseguramiento de la calidad, al no limitarlo a la ausencia de desviaciones sino a una práctica que reduce integralmente el potencial de ocurrencia de las no conformidades sobre la base del conocimiento de las variables del producto y de su proceso productivo (Weinberg, 2011).
Por lo tanto, QbD ha permeado transversalmente todas las áreas del desempeño farmacéutico, encontrándose estudios que incluyen, por ejemplo, el diseño de principios activos, el desarrollo de formas farmacéuticas, su escalamiento y manufactura y, la formulación de metodologías para análisis, limpieza y control en proceso. Esto ha planteado oportunidades y retos en términos de productividad, los que han dado origen a la creación de modelos de manufactura más eficientes en la gestión de los insumos y del capital intelectual (Reklaitis, Khinast y Muzzio, 2010). Al igual que los conceptos de calidad que han precedido a QbD, para los que en diferentes tipos de organizaciones se ha demostrado la relación entre la innovación, el cumplimiento de los requisitos de los productos y la satisfacción de los clientes (Al-Hakim y Jin, 2014), es predecible que la puesta en marcha de programas de aseguramiento de la calidad que involucren QbD como uno de sus elementos esenciales, fortalezca la capacidad innovadora de una empresa.
Para el caso colombiano, la implementación de QbD significaría un aporte para lograr una industria farmacéutica a la vanguardia de las tendencias globales y competitiva en el contexto de la apertura económica que experimenta el país, porque aunque se posiciona como cuarta en Latinoamérica después de Brasil, México y Argentina, y agrupa aproximadamente a 217 empresas, su contribución al valor agregado de la manufactura nacional es limitada y su tasa de apertura exportadora se ha estancado desde el año 2010 (Asociación Nacional de Empresarios de Colombia - ANDI, 2014; Arcila, 2011), lo que se ha atribuido entre otros, a su baja capacidad de innovación relacionada con problemas tecnológicos de diversa índole en el Sistema de Aseguramiento de la Calidad, el área de desarrollo e innovación y el perfil exportador de los laboratorios (Gallo, 2009).
Como un aporte en este sentido, el presente trabajo de revisión analiza en primer lugar las circunstancias que dieron origen a QbD y explica su aplicación en el desarrollo de productos farmacéuticos, articulando de forma didáctica los lineamientos técnicos y las herramientas básicas, para finalmente discutir algunas de sus aplicaciones, beneficios y desacuerdos. A partir de lo anterior, el objetivo del presente trabajo es contribuir al entendimiento de los conceptos asociados a QbD en el sector farmacéutico industrial colombiano. Para tal fin, fueron revisados trabajos de investigación en el área, publicados en revistas académicas tales como Pharmaceutical Engineering, Journal of Pharmaceutical Innovation, International Journal of Pharmaceutics y Pharmaceutical Research. Igualmente, fueron consideradas las guías y lineamientos de las organizaciones internacionales más importantes en el tema, dentro de las que se destacan la ICH y la Food and Drug Administration (FDA).
2. La historia y el contexto de la calidad desde el diseño
La calidad desde el diseño toma elementos desarrollados décadas atrás por diferentes expertos de la calidad y de la gestión del conocimiento y comparte principios con otras metodologías de desarrollo de producto y de gestión de la calidad. Como se presenta en la figura 1, estos elementos han evolucionado hasta lograr una visión holística de la calidad. En sus inicios, la calidad se limitaba a la inspección del producto; sin embargo, fue complementada con el concepto de control de la calidad sistemático y estadístico, consolidando el Control Total de la Calidad (Total Quality Control) de Armand Feigenbaum (Feigenbaum, 1961). Esto permitió una novedosa definición del aseguramiento de la calidad, en donde la documentación se constituye en un aspecto estructural. Igualmente, surgieron estándares internacionales como las normas ISO 9000 (ISO, 2000, 2005, 2008), que hicieron extensiva la calidad a todos los miembros de la empresa con un enfoque centrado en el cliente, los procesos y su integración en sistemas.
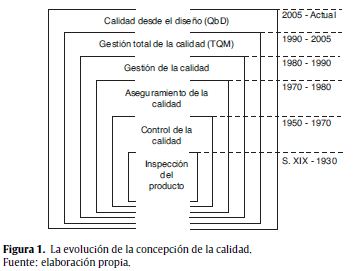
Sobre esta base y los aportes de Edward Deming más todos aquellos provenientes de la teoría administrativa de los años 90, surgió el concepto de Gestión Total de la Calidad (Total Quality Management) buscando satisfacer al consumidor por medio de la mejora continua. Por su parte, Juran (1992) postula la trilogía de la planeación, el control y la mejora de la calidad, afirmando que las desviaciones están relacionadas con la forma como la calidad es concebida desde el principio, es decir desde su diseño, garantizándose y progresando al interior de la empresa gracias a estrategias de control adecuadas y a actividades de mejoramiento continuo. En la práctica, Total Quality Management evidenció la necesidad de trabajar desde la base del entendimiento y la experiencia asociados al producto y a su proceso de fabricación, lo que en la actualidad se conoce como gestión del conocimiento y gestión del riesgo y que se reconocen como los fundamentos de QbD (ICH, 2005, 2009; Organización Mundial de la Salud-OMS, 1996).
De esta forma, el objetivo de la gestión del conocimiento es obtener la información indicada para las personas adecuadas en el momento oportuno, de forma que pueda ser empleada eficientemente (Guebitz, Schnedl y Khinast, 2012). Sobre esta base, se destacan diversos aportes en el entendimiento de la información tanto del producto como de los procesos tales como los de Nonaka y Takeuchi (Nonaka, 1994) sobre la teoría de la creación del conocimiento, que desarrolla las bases de la transformación del conocimiento tácito a explícito y la relación de este con la calidad. Igualmente, se deben considerar los trabajos de Henderson y Clark (Henderson y Clark, 1990) sobre la necesidad de dos tipos de conocimiento en el desarrollo de productos: el de los componentes y el de la interacción entre componentes (arquitectural) y de Chandy y Tellis (1998) acerca de la relación entre conocimiento y desarrollo de nuevos productos considerando la tecnología y el mercado, lo que exige definir un componente de novedad en el producto y el grado de cumplimiento de las necesidades del cliente. Finalmente, resultan de gran utilidad los aportes de Alavi y Leidner (2001) que señalan que el conocimiento puede ser procedimental (know-how), causal (know-why), condicional (know-when) y relacional (know-with), siendo un enfoque pragmático que clasifica el conocimiento de acuerdo con su utilidad en la organización.
En consecuencia, la gestión del conocimiento es un instrumento de ayuda a los diseñadores y productores de medicamentos en la implementación de QbD (Guebitz et al., 2012). Actualmente, la transferencia de conocimiento al interior de las empresas encuentra barreras entre sus departamentos y mucha de la información documentada no es accesible fuera de la función para la que ha sido diseñada e incluso, si tales reportes son accesibles, se hace difícil obtener información adicional a partir de los documentos existentes. Por ello, dada la complejidad de los productos farmacéuticos y las restricciones de presupuesto y de tiempo, se necesita apalancar el conocimiento desde diferentes fuentes como la experiencia de la propia empresa en sus procesos y productos similares y en las etapas previas de desarrollo, así como la experiencia de otras compañías reportada en la literatura.
Igualmente se debe tener en cuenta el conocimiento ganado en las investigaciones de eventos asociados a no conformidades (resultados fuera de especificación, retiros de producto, reclamaciones, etc.). Asimismo se debe reconocer que los avances en tecnología han permitido la generación rutinaria de volúmenes importantes de información, sin embargo, su procesamiento, interpretación y divulgación deben optimizarse por medio del empleo de herramientas de análisis de datos incluyendo aquellos estadísticos, la creación de visualizaciones que faciliten la monitorización de los procesos y el diseño de estrategias para comunicarlos de forma efectiva en la empresa (Rathore, Bansal y Jaspinder, 2013).
De otro lado, como lo presenta la ICH en su guía de calidad Q9, la gestión del riesgo propende por una cultura de previsión y análisis que tiene como soporte el conocimiento adquirido por la empresa acerca de sus productos y procesos. En términos generales, la gestión del riesgo requiere la identificación de los riesgos, su análisis y el diseño de estrategias para evitarlos o mitigarlos. El conocimiento ganado durante la gestión del riesgo debe también administrarse, haciendo énfasis en la comunicación de los resultados, lo que evidencia la estrecha dependencia de las gestiones del riesgo y del conocimiento a la luz de QbD. En adición, debe revisarse periódicamente la eficacia de las estrategias implementadas para evitar o mitigar los riesgos y diseñar las acciones correctivas y preventivas que se consideren pertinentes (ICH, 2005).
Con estos propósitos, la gestión del riesgo dispone de varias aproximaciones metodológicas. Para identificar los riesgos son útiles por ejemplo los análisis de causa-efecto y para la evaluación puede emplearse el Failure Mode Effects and Criticality Analysis (FMECA) desarrollado a finales de los años cuarenta; el Failure Mode and Effect Analysis (FMEA) ampliamente utilizado desde la década de los cincuenta; el Fault Tree Analysis (FTA); el Hazard Operability Analysis (HAZOP) y el Hazard Analysis and Critical Control Points (HACCP) que surgieron en los años sesenta y, el Risk Analysis and Mitigation Matrix (RAMM) propuesto más recientemente. Es así que el Anexo 7 del Informe 37 de la OMS recomienda HACCP como herramienta para asegurar la calidad, resaltando posteriormente su importancia en el Anexo 3 del informe 45, cuando establece que los conceptos de aseguramiento de la calidad, buenas prácticas de manufactura, control de calidad y gestión del riesgo en calidad son aspectos interrelacionados y deben ser responsabilidad de todo el personal de la empresa. Igualmente, haciendo referencia al desarrollo de productos genéricos, en el Anexo 3 del informe 46, se enfatiza en la gestión del riesgo y se proponen lineamientos básicos para el diseño de experimentos enfocados a cumplir el perfil de calidad del producto. Finalmente, el informe 47 en su segundo anexo, establece una guía detallada sobre gestión del riesgo en calidad. En este campo también debe destacarse la guía para el análisis de riesgo y criticidad, Military Standard-1629A, promulgada por el Departamento de Defensa de los Estados Unidos (1980).
En el ámbito farmacéutico, tanto la gestión del conocimiento como la gestión del riesgo han sido la base de diferentes guías técnicas para el desarrollo del producto, el aseguramiento de la calidad y la productividad. Estas han catalizado progresivamente la evolución de la industria farmacéutica desde la producción en masa y poco flexible hacia la innovación en productos y procesos, evidenciada hoy por hoy en la solución de retos como la personalización de tratamientos farmacológicos, el control de procesos productivos en tiempo real y la demanda variable en el consumo de medicamentos (Politis y Rekkas, 2011), (fig. 2). Como soporte de esta evolución se encuentran las guías ICH Q8 sobre desarrollo farmacéutico (ICH, 2009), ICH Q9 que trata sobre la gestión del riesgo en calidad (ICH, 2005) e ICH Q10 que presenta el sistema de calidad farmacéutico (ICH, 2008). Asimismo, se destacan las guías sobre tecnología de análisis en proceso y gestión del riesgo promulgadas por la FDA (2002, 2004), las guías sobre implementación de la calidad en el ciclo de vida del producto de la International Society for Pharmaceutical Engineering, los lineamientos para la aplicación de la verificación continua de la calidad en la manufactura farmacéutica y biofarmacéutica ASTM E2537 de la American Society for Testing and Materials (ASTM, 2008) y algunos anexos de los informes del Comité de Expertos de la Organización Mundial de la Salud en Especificaciones para las Preparaciones Farmacéuticas (OMS, 2012, 2013).
De acuerdo con lo anterior y a manera de síntesis, QbD interrelaciona dos conceptos de gestión fundamentales: riesgo y conocimiento. La gestión del riesgo permite identificar y valorar las fuentes de no conformidades optimizando el empleo de recursos en sistemas robustos de calidad. Al integrar la gestión del conocimiento, se logra la mejora continua de los productos satisfaciendo las demandas del cliente, que es el principal propósito de la gestión de la calidad (Ansari, Khobreh, Nasiri y Fathi, 2009) mientras permite desarrollar un conjunto definido de competencias esenciales, catalizando el desarrollo y la introducción de nuevos productos en el mercado (Somma, 2007). En consecuencia, la implementación de QbD provee un mayor nivel de aseguramiento en la calidad del producto, disminuciones de costo y una productividad mejorada para la industria, facilitando a su vez una eficiente vigilancia por parte de los organismos reguladores (Riley y Li, 2011).
3. La calidad desde el diseño en la industria farmacéutica
En el entorno farmacéutico, QbD se ha consolidado gracias a la coincidencia de cuatro factores (Kamm, 2007). En primer lugar, la disponibilidad de guías sobre desarrollo farmacéutico y sobre la aplicación del concepto de criticidad para clasificar la aceptabilidad de las variaciones (ICH, 2009). En segundo lugar, la evolución del concepto de vigilancia de organismos como la FDA hacia un modelo autorregulado donde la función de aseguramiento de la calidad de cada empresa actúe como la agencia regulatoria primaria. En tercer lugar, la estadística y la tecnología informática han desarrollado herramientas sistemáticas basadas en la interpolación de datos empíricos con un poder predictivo robusto. Finalmente, en cuarto lugar, la aceptación por parte de las entidades regulatorias a nivel mundial, de la toma de decisiones basadas en el riesgo impulsadas por la FDA en 2001 (FDA, 2002), lo que hace posible monitorear los procesos sin depender del control de todas las variables concebibles en la manufactura del producto terminado.
Para promover la implementación de QbD en el sector industrial farmacéutico colombiano, el primer paso consiste en entender tanto su terminología como su metodología. Para ello, el presente artículo explica QbD desde la óptica del diseño, evidenciando cómo la calidad se construye en el desarrollo de un producto farmacéutico al responder a las preguntas ¿qué se quiere alcanzar? y ¿cómo se puede alcanzar? Dichas preguntas permean los cuatro dominios de la teoría del diseño axiomático (dominio del consumidor, dominio funcional, dominio físico y dominio del proceso) en donde se busca analizar la transformación de las necesidades del cliente en requerimientos funcionales, parámetros de diseño y variables de proceso. Se debe tener en cuenta durante el desarrollo de productos la importancia de los cuatro dominios, constituyentes del proceso creativo que conlleva un diseño, además del conocimiento sobre los materiales y sus procesos de transformación con los que debe contar un diseñador (Vallejo, Cortés, Espinosa y Barbosa, 2004).
A diferencia del diseño basado en QbT, QbD busca disminuir la rigidez y la dependencia en las especificaciones que evalúan el desempeño de un producto a través de sus datos históricos. Esto facilita la identificación de los parámetros críticos del proceso y sus límites de control en la etapa de desarrollo y contribuye a diseñar un producto que cumpla consistentemente con los atributos críticos de calidad previamente determinados. Además, al predecir eficientemente el impacto de las variables del proceso en las especificaciones del producto, es posible un mejor aseguramiento de la calidad del medicamento (Staples, 2010).
En este sentido, varios autores han propuesto métodos sistemáticos de aplicación de QbD basados en las guías de la ISPE, la ICH y la FDA, los cuales poseen algunos elementos comunes y otros complementarios (Lionberger, Lee, Lee, Raw y Yu, 2008; Roy, 2012; Yu, 2008; Nasr, 2006). Así, como se presenta en la figura 3, es posible integrar la mayoría de las propuestas en cuatro pasos: a) identificación de objetivos, b) establecimiento de requerimientos, c) construcción del diseño y d) articulación del proceso en el marco del mejoramiento continuo. A continuación se presenta en detalle cada una de estas etapas, empleando un símil práctico a través de un deporte como el golf. Igualmente se exponen las herramientas y los principios más importantes en cada fase y se explica su articulación en el marco de la mejora continua.
3.1 Etapa de identificación de objetivos
De acuerdo con la definición de QbD dada por la ICH, el primer paso en su implementación es 'partir de objetivos predefinidos' teniendo en cuenta las necesidades reales del cliente. Con ello, todas las etapas subsecuentes tendrán un sentido congruente y se logrará un concepto de calidad que va más allá de la 'aptitud para el uso'.
A manera de ejemplo, se debe suponer que el diseñador del medicamento y de su proceso de fabricación puede compararse con un golfista, cuyo objetivo al que debe direccionar los golpes se encuentra a una distancia considerable. En adición, los hoyos a los que apunta son bastante pequeños y difíciles de ubicar en todo el campo. El golfista no entiende el efecto del viento, el ángulo y la fuerza del golpe sobre las bolas, ni ha seleccionado su palo según las características óptimas para darle fuerza y precisión a su disparo. Además, presenta problemas visuales, por lo que le es difícil saber cómo están llegando las bolas al objetivo. Tampoco recuerda las técnicas que emplea y su eficacia. Lo único que hace es, tras una ronda de golpes, acercarse al área de impacto y observar cuántas veces se aproximó al pequeño objetivo en una forma casi aleatoria. Intenta perfeccionar su procedimiento al repetir constantemente los golpes. Es así que el golfista trabaja bajo un enfoque QbT. Dado lo anterior, cansado de fallar, se empeña en incrementar su tasa de éxito a través de QbD, ubicando en un mapa del campo la localización de los blancos (las necesidades del cliente) y las dimensiones del verde, es decir hasta dónde es aceptable que fluctúe la llegada de las bolas sin que se malgaste el golpe. Igualmente, adquiere algunos libros sobre golf, estudia los reglamentos y empieza a analizar lo que ha estado haciendo hasta el momento. En términos de QbD en la industria farmacéutica, lo que realizó fue lo siguiente:
- Establecer el perfil del producto objetivo (Target Product Profile), el cual es construido a partir de las necesidades explícitas del consumidor y de los requerimientos reguladores que debe cumplir el producto. El Target Product Profile es entendido como aquella información que se encontrará en la etiqueta del medicamento como por ejemplo la forma farmacéutica, la dosis, el principio activo, la estabilidad, etc. Igualmente se tiene en cuenta al consumidor secundario, es decir quien formula y administra el producto.
- Definir el perfil de calidad del producto objetivo (Target Product Quality Profile), que se refiere a los atributos cuantitativos para lograr la eficacia y seguridad deseadas para el medicamento: contenido de impurezas, ensayo de disolución, valoración, etc.
- Recopilar información sobre el principio activo, los excipientes y el proceso, generando un espacio de conocimiento, es decir, el estado del arte que relaciona la mayor cantidad de datos preexistentes, estableciendo un punto de partida que oriente la planeación del desarrollo. Esto puede complementarse con un análisis de riesgo para encontrar aquellos requerimientos de información que deben ser resueltos con investigación posterior.
3.2 Etapa de establecimiento de los requerimientos
Ahora, el golfista entiende que no es necesario impactar exactamente en el hoyo y que algunos agujeros son más críticos que otros. Por tal razón, prioriza los blancos con mayor puntuación y se hace una idea de hasta dónde pueden caer las bolas, maximizando su puntuación con la menor cantidad de golpes y no con uno solo. También empieza a preocuparse por el material y forma de sus palos e identifica los factores que afectan su desempeño (velocidad y dirección del viento, fuerza de golpe, ángulo de golpe, tensión muscular, entre otros) analizando el riesgo que trae el pasar por alto cada uno de esos factores. Adicionalmente, estudia cada una de las exigencias realizadas en el reglamento del juego con el propósito de tenerlos presentes en la práctica. En los términos del QbD, lo que hizo se entiende de la siguiente manera:
- Determinar los atributos críticos de calidad (Critical Quality Attribute [CQA]) definidos como aquellas características que después de un análisis de riesgo resultan ser necesariamente controladas. Estas se refieren usualmente al producto terminado, aunque pueden encontrarse en la materia prima y en los productos intermedios, teniendo como eje de decisión (criticidad) los requerimientos establecidos para los clientes internos y externos del proceso.
- Identificar los atributos críticos del material (Critical Material Attribute [CMA]) con una evaluación de riesgo, de manera que sobre una base científica, se establezca el impacto de las propiedades de los componentes de la formulación en el cumplimiento de los CQA.
- Identificar los parámetros críticos del proceso (Critical Process Parameter [CPP]) y las fuentes de variación. Nuevamente su selección debe partir de un análisis de riesgo complementado con una verificación experimental de los parámetros priorizados.
El principal concepto de esta etapa es la 'criticidad', que es empleada para describir los atributos de una materia prima, de un producto terminado o del proceso de fabricación de un medicamento. El criterio para calificar los atributos como críticos resulta del análisis de riesgo, siendo válido únicamente si se dispone de un verdadero conocimiento del producto, del entendimiento de los procesos y de la experiencia (Garcia, Cook y Nosal, 2008). Sin embargo, el uso de la palabra 'crítico' para definir un atributo suele ser subjetivo, pues se contextualiza en la evaluación circunstancial del riesgo relativo, basándose en el conocimiento empírico, en la experiencia directa o en datos. Por lo tanto, su significado no es ni universal ni definitivo (Nosal y Schultz, 2008).
En la práctica, la valoración del riesgo busca responder cuatro preguntas fundamentales: ¿qué podría salir mal?, ¿cuál es la probabilidad de que salga mal?, si sale mal, ¿cuál es el impacto y su gravedad?, y si sucede, ¿se detectará la falla? Así, el nivel relativo o grado de riesgo es evaluado con relación a la probabilidad de ocurrencia y a la capacidad para detectar la falla e impedir un daño potencial para el producto o el consumidor o el incumplimiento de una exigencia regulatoria (Nosal y Schultz, 2008). Por lo tanto, es preciso enfatizar que la valoración del riesgo, es decir su identificación, análisis y evaluación, es una etapa dentro de la gestión del riesgo, en adición al control, la comunicación y la revisión. En las fases iniciales del ciclo de vida del producto, el resultado de la evaluación inicial del riesgo probablemente hará evidente la necesidad de una investigación experimental adicional para caracterizar las fuentes de riesgo potenciales que generan impacto en los CQA del producto (Guebitz et al., 2012).
Dependiendo de la importancia de los procesos, el análisis del riesgo puede tener diferente exigencia, lo que permite clasificarlo como simple (utilizando por ejemplo un árbol de decisión de fallas-FTA), moderado (en el que se aplican herramientas como HAZOP y RAMM) o detallado (que requiere el empleo de FMEA o HACCP, por ejemplo), (Brindle, Davy, Tiffany y Watts, 2012). Al identificar un riesgo significativo, es deseable su eliminación. Sin embargo, en la mayoría de los casos es imposible eliminar el riesgo en su totalidad resultando un 'riesgo residual' que debe ser aceptado y controlado para evitar que se convierta en una fuente de variación significativa de la calidad de los productos. Si se encuentra un riesgo inaceptable, se selecciona un nuevo diseño para el producto o proceso o se abandona el producto (Hulbert, Feely, Inman, Johnson, Michaels, Mitchell y Zour, 2008).
3.3 Etapa de construcción del diseño
Después de estudiar cuidadosamente los factores de éxito -o de fracaso-, es el momento de practicar los golpes antes de una verdadera competición. Por eso, el golfista 'experimenta' con diferentes técnicas de golpe, de la forma más eficiente posible. Con este propósito, varía los factores identificados como críticos entendiendo empíricamente cómo están afectando los resultados. Así, establece un conjunto de estrategias según sus habilidades particulares, que aplicará a diversas situaciones garantizando eficiencia y puntería. En su mente define paso a paso la estrategia y su entrenamiento permanente le permite optimizarla. En este sentido, en el diseño farmacéutico lo que se realizó fue:
- Diseñar un plan experimental para establecer las correlaciones necesarias entre los CQA y las fuentes de variación en la formulación, desarrollando un prototipo de producto.
- Seleccionar preliminarmente el proceso de manufactura, detallando sus operaciones unitarias y la articulación entre las entradas y las salidas en cada paso.
- Combinar la información disponible en la definición de los espacios de diseño del proceso por medio del diseño experimental adecuado.
En general, los procesos de manufactura en la industria farmacéutica se ven influenciados por una matriz compleja de parámetros de entrada y de salida, incluyendo los CMA y Critical Process Parameter, que pueden estar interrelacionados o ser independientes entre sí. El estudio de tales parámetros de entrada y su relación con los de salida, permite establecer de forma práctica y eficiente los espacios de diseño (Design Space [DS]). Según la ICH, un DS corresponde a la combinación multidimensional e interactiva de las variables de entrada y los parámetros de proceso que demuestran proveer un adecuado nivel de calidad, es decir, la variación que se permite para las variables de entrada (como por ejemplo los atributos de los materiales o las condiciones de operación) garantizando el cumplimiento de las especificaciones establecidas para las variables de salida (por ejemplo, una característica de calidad del producto). Esto se logra por medio de la exploración del espacio operacional de todas las entradas seleccionadas con respecto al comportamiento de las variables de salida, es decir, toda la información preexistente, adquirida o generada, acerca de la respuesta de un proceso o material frente al cambio de las variables de un sistema, consolidando conocimiento explícito, cuantitativo o cualitativo, que asegure con suficiencia la calidad del producto (Lepore y Spavins, 2008). En este contexto, el DS es identificado como la región robusta dentro del espacio de conocimiento que permite la correcta manufactura del producto dentro de una región del espacio operacional, comprendido como todos los valores posibles de la variable de entrada en una operación (fig. 4).
En la práctica, el establecimiento de un DS se basa en el uso de experimentos para una o varias operaciones donde la relevancia de los parámetros de proceso está siendo investigada. Para tal fin, la aplicación de una estrategia basada en el diseño estadístico de experimentos (Design of Experiments [DoE]) permite evaluar los efectos de diferentes parámetros del proceso y de los atributos del material sobre los CQA del producto, lo que en el trabajo experimental significa un ahorro de recursos entre cuatro a ocho veces más que aplicando un modelo experimental de un factor a la vez (Torbeck y Branning, 2009). Existen diferentes modelos de DoE tales como Plackett-Burman, Taguchi y Box-Behnken (de superficie), cuya correcta selección es importante para el éxito del experimento y requiere considerar el número de variables e interacciones a estudiar, la complejidad del diseño experimental, la validez estadística y la efectividad del modelo, la facilidad de interpretación e implementación de la información generada dada su complejidad, la naturaleza del problema y las restricciones de recursos (Savic, Marinkovic, Tasic y Krajnovic, 2012). No obstante, independientemente del modelo empleado, es fundamental que el DoE se base en una revisión sistemática de literatura técnica especializada y en el conocimiento previamente generado por la empresa.
Al establecer un DS, es fundamental demostrar que los parámetros no considerados en el diseño experimental, son parámetros no críticos y no interactúan significativamente con los CQA. Estos parámetros se deben reducir a un mínimo, de esta manera, cuando se demuestra que una variable es 'no-interactuante', se pueden establecer especificaciones sin rangos, las que en conjunto con los DS constituyen la estrategia de control de manufactura (Juran, 1992; Lionberger et al., 2008; Shivhare y Mccreath, 2010). Sobre esta base, al momento de solicitar una autorización de comercialización, el DS puede ser considerado como una fotografía instantánea del conocimiento del proceso. Sin embargo, el DS continúa evolucionando a medida que se genera nuevo conocimiento durante la fabricación rutinaria del producto, lo que puede conducir a cambios postaprobación (Garcia et al., 2008).
Aunque el concepto de DS resulta novedoso, en realidad es comúnmente empleado en la práctica de fabricación industrial de medicamentos. Un ejemplo de una situación en la que se emplea sin que sea evidentemente conocido por quienes ejecutan las acciones de calidad, es cuando una materia prima se encuentra fuera de los límites aceptables de entrada (CMA). En este caso, de acuerdo con los resultados de control de calidad de la materia prima, los responsables del proceso según su experiencia y conocimiento, modifican algunas condiciones de la fabricación para compensar el efecto del cambio observado en la calidad del material de partida sobre la calidad del producto (McGregor y Bruwer, 2008). Cuando este conocimiento se transforma en explícito por medio del DoE, se genera el espacio de conocimiento anteriormente expuesto y cuando se limita con los rangos del CQA, se define el DS.
3.4 Etapa de articulación del proceso
Finalmente, el golfista emplea su tiempo en desarrollar habilidades para determinar con mayor precisión cómo están cambiando las variables que afectan la eficacia de sus golpes, observando la evolución de su desempeño. Adicionalmente, emplea su experiencia para crear conocimiento aplicable y extrapolable a diferentes situaciones del juego, de manera que logre estar preparado para afrontar diversos escenarios. Nuevamente, poniendo esta situación en términos de QbD, se encuentra que lo que se realizó fue:
- Estandarizar el proceso de manufactura teniendo presente el cumplimiento de los CQA.
- Entender y asimilar la interacción del conocimiento generado para valorar correctamente el impacto de los componentes de la formulación y los parámetros de proceso en la calidad.
- Establecer una estrategia de control incluyendo controles a los materiales de entrada, monitorización y control al proceso, creación de DS para una o varias operaciones o realización de pruebas al producto terminado. En esta etapa suelen integrarse estrategias de tecnología de análisis en proceso (Process Analytical Technology [PAT]) para liberar productos en tiempo real e incrementar el nivel de confianza en su calidad.
- Validar el proceso, es decir que el conocimiento generado durante el desarrollo del producto y del proceso sustente por sí mismo los requerimientos de la validación.
Considerando que uno de los objetivos de QbD es mantener el proceso bajo control para garantizar el cumplimiento de los atributos deseados del producto, PAT adquiere relevancia. Esta tecnología implica el análisis de los atributos de calidad durante las diferentes etapas de manufactura, permitiendo realizar ajustes en tiempo real a los parámetros del proceso para obtener el perfil de producto deseado en cada etapa de la transformación (Mhatre y Rathore, 2009).
Generalmente los controles realizados en la manufactura farmacéutica se clasifican como: in-line o controles que determinan el comportamiento de una propiedad directamente sobre el flujo de proceso; off-line o controles realizados en laboratorios conexos sobre muestras extraídas del sistema; at line o controles realizados al pie del proceso de manera semiautomática y on line o controles automatizados en tiempo real. Estos últimos son sistemas de control que ajustan la variable respuesta directamente sobre el flujo del proceso. Un sistema de producción que emplee PAT puede integrar estos cuatro tipos de controles (Wu, Khan y Hussain, 2007). En los últimos años, la implementación de PAT se ha direccionado hacia el desarrollo de sensores on-line y sondas que permitan el control en proceso (Wu et al., 2007). Algunos ejemplos de herramientas utilizadas en el control de procesos en línea para operaciones de la industria farmacéutica incluyen: infrarrojo mediano, espectroscopía raman, espectroscopía de infrarrojo cercano (NIR) y mediciones de microscopía confocal de reflectancia en línea para la cristalización de principios activos; espectroscopía NIR, raman y espectroscopía de fluorescencia inducida por láser para la operación de mezcla; mediciones in-line/at-line de la distribución del tamaño de partícula y NIR para mediciones de humedad en el gránulo en la operación de granulación con equipos de alto corte; NIR in-line para el contenido de humedad y mediciones at-line del tamaño de gránulo en operaciones realizadas en lecho fluidizado; sensores de fatiga en los componentes sujetos a presión de las tableteadoras y mediciones at-line de contenido de principio activo acoplados a determinadores del peso de tabletas en la operación de compresión para ofrecer resultados instantáneos del ensayo de valoración (Troup y Georgakis, 2013).
3.5 Consolidación de la calidad desde el diseño y mejora continua
Cuando la experiencia y la práctica constante de su deporte le han significado al golfista el reconocimiento de sus habilidades y experticia, este se consolida como un referente para sus pares. Su autoridad en la materia le permite ser el entrenador de nuevos talentos o quizás, documentar su trayectoria en un libro práctico para aquellos talentos o tal vez, documentar su trayectoria para aquellos también interesados en el golf, estando siempre atento a los nuevos desarrollos en la técnica y las tendencias del deporte. En QbD, para lograr su fortalecimiento en una empresa, es necesario realizar las siguientes actividades de forma transversal durante todo el ciclo de vida del medicamento:
- Transformar el conocimiento que se considera tácito en explícito y emplear las herramientas de gestión del conocimiento para administrarlo, permitiendo la mejora continua y la retroalimentación a través de la experiencia.
- Monitorización y actualización permanente del diseño y de la manufactura para asegurar la consistencia del mismo dentro de la filosofía del mejoramiento continuo.
Es importante resaltar que todas las etapas asociadas a QbD convergen en el desarrollo de una estrategia de control, relacionada directamente con el nivel de entendimiento del proceso. Por ello dicha estrategia evoluciona a medida que incrementa la experiencia en la manufactura de manera que puede ser modificada, removida o complementada con nuevos controles. Así, una vez que el producto ha sido desarrollado y transferido para la producción a escala industrial, se genera información que hace necesaria la aplicación de nuevas evaluaciones del riesgo a la profundidad pertinente debido a que se dispone de otras fuentes que ofrecen oportunidades para identificar riesgos, como por ejemplo el sistema de monitorización del desempeño y de la calidad o el sistema de manejo de acciones correctivas y preventivas (Potter y Berridge, 2010).
Es necesario entonces, realizar revisiones continuas de las evaluaciones de riesgo y los planes de mitigación para demostrar que la estrategia de control es la adecuada, basándose en la historia de manufactura del producto (Davis, Lundsberg y Cook, 2008). De acuerdo con esto, se resalta que el concepto de mejoramiento continuo está estrechamente ligado a QbD y fortalece el espíritu de la calidad en todo el ciclo de vida del producto. En este sentido, algunos ejemplos de mejora continua incluyen el ajuste de las condiciones de operación, la introducción de técnicas avanzadas de control como mediciones en línea, la implementación de nuevos equipos, el rediseño de alguna etapa del proceso, la puesta en marcha de proyectos de manufactura esbelta, la simplificación de la documentación, la automatización del proceso, la eliminación de alguna operación unitaria o simplemente, el cambio en el espacio de diseño propuesto inicialmente (Trivedi, 2012).
4. Los efectos de la calidad desde el diseño: oportunidades para la industria farmacéutica
La tabla 1 sintetiza la literatura técnica disponible acerca de la implementación de QbD en el campo farmacéutico. Como se observa, QbD ha sido aplicado al diseño de productos, al estudio y optimización de procesos productivos y al desarrollo de operaciones de apoyo como por ejemplo, la limpieza, el desarrollo de metodologías analíticas y el desarrollo de software. Según Janeth Woodcock, Directora del Centro para la Investigación y Evaluación de Medicamentos de la FDA, el resultado de implementar QbD es la maximización de la eficiencia, de la agilidad y de la flexibilidad del sector de manufactura farmacéutica, produciendo con un elevado grado de confianza, medicamentos de calidad sin la necesidad de una vigilancia intensiva (Woodcock, 2005). Aclarando algunas concepciones sobre el tema, el ex Director de la Oficina de Evaluación de la Calidad de Nuevos Medicamentos de los Estados Unidos, Moheb Nasr, explica que el principal beneficio de QbD no es la flexibilidad regulatoria, sino el potenciar la calidad de los medicamentos y la eficiencia de su desarrollo y manufactura, representando beneficios en términos de costos para la industria, posiblemente ventajosos para el consumidor. La motivación es hacer lo correcto a partir de bases científicas, enfocándose al riesgo y asegurando que se logren medicamentos de alta calidad, seguros y efectivos (Hall y Runas, 2007).
De otro lado, QbD ofrece beneficios para las empresas en términos de flexibilidad, tales como facilitar el desarrollo de la producción en diferentes plantas, modificar las escalas para satisfacer la demanda y llevar a cabo procesos con una amplia gama de equipos sin incidir en la calidad final del producto. También se ha aprovechado el incremento de la seguridad en el desarrollo del producto para promover el aprendizaje e introducir cambios en la cultura organizacional, que se manifiestan por ejemplo, en que los empleados en el área de manufactura logran entender mejor las necesidades de sus clientes internos. De igual forma QbD provee otros beneficios que son difíciles de cuantificar, tales como el mejoramiento de la imagen corporativa, el consenso en el lenguaje de la producción al interior de la compañía y la posibilidad de compartir las mejores prácticas en el diseño de producto, el aseguramiento de la calidad, la gestión de la manufactura y la gestión del conocimiento (FDA, 2009). Adicionalmente, QbD ha sido empleada para lograr un mejor entendimiento de las guías y exigencias regulatorias facilitando la interacción con los organismos de control (Hulbert et al., 2008). Frente a este último aspecto, los organismos reguladores son grandes beneficiarios, pues el origen de la implementación de QbD provino de las mismas agencias estatales, particularmente de la FDA. En primer lugar, las solicitudes de autorización de comercialización a revisar se vuelven más claras y concretas puesto que se soportan en la ciencia del producto aportando evidencia altamente confiable. Igualmente, se presentan menos acciones correctivas durante el ciclo de vida del producto, lo que les permite una gestión más ágil (Staples, 2010).
Sin embargo, a pesar de la experiencia y evidencia que se dispone en la actualidad respecto a los beneficios de implementar QbD, la industria aún no la ha apropiado ampliamente aduciendo razones que varían desde la falta de conocimiento acerca de cómo incorporarla en su práctica diaria y las dificultades desde el punto de vista técnico asociado a la producción farmacéutica, hasta la falta de compromiso en la compañía debido a que los beneficios económicos de la mejora de la calidad no son bien entendidos y generalmente se estiman en el corto plazo (Maes y Van Liedekerle, 2006). A este respecto, es importante ser conscientes de que los beneficios de la inversión en QbD pueden visibilizarse solo en el mediano plazo (en la fase de posmercadeo) (Savic et al., 2012), principalmente en la reducción entre un 15% a un 30% de los costos de calidad (DeFeo, 2001).
Algunas compañías se resisten al cambio argumentando un incremento en los costos, el tiempo y la burocracia para el desarrollo del producto. El posible aumento en el tiempo de desarrollo de producto, resulta problemático especialmente para los laboratorios fabricantes de genéricos, por restricciones de tiempo. En adición, la implementación de QbD podría también ser difícil de manejar cuando la elaboración del medicamento se hace por subcontratación debido a que los responsables del desarrollo del producto y del proceso no pueden integrarse fácilmente con quienes responden por la fabricación del medicamento (Drakulich y Van Arnum, 2010). En lo relacionado con PAT, una herramienta valiosa para QbD, dado que fue desarrollada a mediados de los años cincuenta por la industria química, fue concebida en medio de un contexto muy diferente a los riesgos y a la alta regulación del sector farmacéutico (Torbeck y Branning, 2009). En una encuesta realizada a diferentes representantes de la industria farmacéutica, se encontró que el 39% de los participantes estaba preocupado por la falta de claridad en el significado del concepto, el 15% tenía reservas sobre la duración de la implementación, el 13% señaló restricciones de presupuesto, el 13% adujo falta de soporte de gestión, el 11% manifestó dificultad de cuantificar los beneficios y finalmente, el 9% señaló la falta de personal calificado para el proceso (Torbeck y Branning, 2009).
En Colombia, desde la experiencia de la academia apoyando a la industria farmacéutica en la implementación de buenas prácticas de manufactura, se reconoce que la validación, sea de procesos, metodologías analíticas, limpieza o sistemas automatizados, ha contribuido de forma significativa para garantizar el cumplimiento de dichos estándares de aseguramiento de la calidad. De acuerdo con lo expuesto en el presente artículo, esta es la esencia de QbD. Sin embargo, aún se requiere entender y apropiar algunos de los conceptos básicos así como las diferentes estrategias que garantizan la adecuada gestión del conocimiento y del riesgo de manera que se logre su implementación exitosa. En ese sentido, este trabajo ofrece un aporte al respecto, particularmente en lo relacionado con el marco conceptual y metodológico de QbD.
5. Conclusiones
Desde principios del siglo XX, QbD ha venido gestándose con el desarrollo del control estadístico de la calidad y se ha consolidado con la implementación de la gestión total de la calidad. Para el caso particular de la industria farmacéutica, garantizar la calidad desde el momento mismo del desarrollo de los productos representa el motor del cambio hacia un verdadero manejo de la gestión del conocimiento y del riesgo. Esto ofrece oportunidades de proyección en las áreas de ingeniería, ciencias básicas y ciencias administrativas, por medio de la investigación de las operaciones, los materiales y los procesos específicos del trabajo farmacéutico. Aunque varios autores han propuesto diferentes metodologías con variaciones en la forma de integrar los pasos conceptuales para llegar a la implementación de QbD, en el presente trabajo la mayoría de ellas se han condensado en cuatro etapas: la definición de objetivos, el diseño del producto y del proceso, la consolidación de QbD y, el control del riesgo y mejoramiento continuo. La evidencia disponible a partir de las experiencias reportadas en la literatura acerca de la implementación de QbD, sugiere que además de asegurar la capacidad de una empresa para proveer medicamentos cada vez con mejor calidad y seguridad para el paciente, se logran beneficios tales como la maximización de la eficiencia, mayor agilidad de los procesos y flexibilidad del sector de manufactura farmacéutica. No obstante, algunas empresas son renuentes a la adopción de QbD, debido principalmente al desconocimiento de los conceptos y de los aspectos metodológicos requeridos y a la percepción de que su puesta en práctica requiere un elevado costo en términos de capital y de tiempo.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Bibliografía
Adam, S., Suzzi, D., Radeke, C. y Khinast, J. (2011). An integrated Quality by Design (QbD) approach towards design space definition of a blending unit operation by Discrete Element Method (DEM) simulation. European Journal of Pharmaceutical Science, 42(1-2), 106-115. [ Links ]
Al-Hakim, L. y Jin, C. (2014). Quality innovation: Knowledge, theory, and practices. Hershey, PA: IGI Global. [ Links ]
Alavi, M. y Leidner, D. (2001). Review: Knowledge management and knowledge management systems: Conceptual foundations and research issues. MIS Quarterly, 25(1), 107-133. [ Links ]
Andreasen, B., Blasi, J., Fabritz, H., Feldthusen, H., Guldager, N. y Moelgaard, G. (2011). Risk-MaPP, ICH Q9, ASTM 2500 in action: project advantages of practical quality risk management approaches. Pharmaceutical engineering, 31(1), 36-43. [ Links ]
ANDI (2014). Presentación cifras de la industria a Mayo de 2014. Cámara de la industria farmacéutica. [consultado 31 Ago 2014]. Disponible en: http://es.scribd.com/doc/202399419/3-Panorama-Actual-de-la-Industria-Farmaceutica-2011-20120321-021943 [ Links ]
Ansari, F., Khobreh, M., Nasiri, S. y Fathi, M. (2009). Knowledge Management Support for Quality Management to Achieve Higher Customer Satisfaction. Proceedings of the 2009 IEEE International Conference on Electro/Information Technology, IEEE EIT 2009, Windsor, Ontario, Canada, Junio 7-9, 2009. IEEE Press, 78-83. [ Links ]
Arcila, R. (2011). De dónde venimos y para dónde vamos en el mercado farmacéutico en Colombia: retos en el corto y largo plazo. Presentación del Director Ejecutivo de la cámara farmacéutica de la ANDI. ANDI. [consultado 31 Ago 2014]. Disponible en: http://www anif.co/sites/default/files/uploads/Rodrigo%20Arcila%20-%20Andi.pptx. [ Links ]
ASTM (2008). ASTM E2537 - Standard guide for application of continuous quality verification to pharmaceutical and biopharmaceutical manufacturing. West Conshohocken, PA: ASTM International. doi:10.1520/E2537-08. [ Links ]
Bos, T., Irving, P. y Rees, P. (2010). Risk-based MES implementation using Hazard. Analysis and critical control points (HACCP). Pharmaceutical engineering, 30(6), 8-22. [ Links ]
Brindle, A., Davy, S., Tiffany, D. y Watts, C. (2012). Risk Analysis and Mitigation Matrix (RAMM) - A risk tool for quality management. Pharmaceutical engineering, 32(1), 26-35. [ Links ]
Campbell, I. y Thibeault, D. (2012). A quality risk management approach to review and monitor cleaning processes. Pharmaceutical engineering, 32(1), 48-55. [ Links ]
Caraballo, I. (2009). Critical points in the formulation of pharmaceutical swellable controlled release dosage forms Influence of particle size. Particuology, 7(6), 421-425. [ Links ]
Chandy, R. y Tellis, G. (1998). Organizing for radical product innovation: The overlooked role of willingness to cannibalize. Journal of Marketing Research, 35(4), 474-487. [ Links ]
Charoo, N., Shamsher, A., Zidan, A. y Rahman, Z. (2012). Quality by design approach for formulation development: a case study of dispersible tablets. International Journal of Pharmaceutics, 423(2), 167-178. [ Links ]
Davis, B., Lundsberg, L. y Cook, G. (2008). PQLI Control strategy model and concepts. Journal of Pharmaceutical Innovation, 3(2), 95-104. [ Links ]
DeFeo, J. (2001). The tip of the Iceberg. Quality Progress, 34(5), 29-37. [ Links ]
Diaz, R., Fernández, G. y Muzzio, C. (2011). Practical application of quality risk management to the filling process of betamethasone injections. Pharmaceutical engineering, 31(4), 84-89. [ Links ]
Departamento de Defensa de los Estados Unidos (1980). Military Standard 1629A. Procedures for performing a failure mode, effects and criticality analysis. Washington D.C. [consultado 11 Sep 2014]. Disponible en: https://src.alionscience.com/pdf/MIL-STD-1629RevA.pdf [ Links ]
Drakulich, A. y Van Arnum, P. (2010). Applying QbD to excipient formulation and development. Pharmaceutical Technology, 34(5), 24-29. [ Links ]
Dubey, A., Boukouvala, F., Keyvan, G., Hsia, R., Saranteas, K., Brone, D., et al. (2012). Improvement of tablet coating uniformity using a quality by design approach. AAPS PharmSciTech, 13(1), 231-246. [ Links ]
Fahmy, R., Kona, R., Dandu, R., Xie, W., Claycamp, G. y Hoag, S. (2012). Quality by design I: Application of failure mode effect analysis (FMEA) and Plackett-Burman design of experiments in the identification of main factors in the formulation and process design space for roller-compacted ciprofloxacin hydrochloride immediate-release tablets. AAPS PharmSciTech, 13(4), 1243-1254. [ Links ]
Feigenbaum, A. (1961). Total quality control. New York, NY: McGraw Hill. [ Links ]
Food and Drug Administration (2002). Pharmaceutical quality for the 21st century: a risk-based approach. Rockville, MD: Department of Health and Human Services - Food and Drug Administration. [ Links ]
Food and Drug Administration (2004). Guidance for industry. PAT-a framework for innovative pharmaceutical development, manufacturing and quality assurance. Rockville, MD: Department of Health and Human Services - Food and Drug Administration. [ Links ]
Food and Drug Administration (2009). Understanding challenges to quality by design. FDA Understanding Challenges to QbD Project [consultado 15 Nov 2014]. Disponible en: http://www.pharmaqbd.com/wp-content/uploads/2011/05/Understanding-Challenges-to-Quality-by-Design.pdf [ Links ]
Gallo, J. (2009). Estudio de la relación proveedor - productor en la gestión de materiales del sector farmacéutico industrial productivo (sfip) de la ciudad de Bogotá. Disertación tesis de Maestría. Bogotá: Universidad Nacional. [ Links ]
Garcia, T., Cook, G. y Nosal, R. (2008). PQLI key topics - criticality, design space, and control strategy. Journal of Pharmaceutical Innovation, 3(2), 60-68. [ Links ]
Guebitz, B., Schnedl, H. y Khinast, J. (2012). A risk management ontology for qualityby- design based on a new development approach according GAMP 5.0. Expert Systems with Applications, 39(2012), 7291-7301. [ Links ]
Hall, G. y Runas, R. (2007). JPI Interviews Moheb Nasr, PhD. Journal of Pharmaceutical Innovation., 2(3-4), 67-70. [ Links ]
Henderson, R. y Clark, K. (1990). Architectural innovation: The reconfiguration of existing product technologies and the failure of established firms. Administrative Science Quarterly, 35(1), 9-22. [ Links ]
Huang, J., Kaul, G., Cai, C., Chatlapalli, R., Hernandez-Abad, P., Ghosh, K., et al. (2009). Quality by design case study: an integrated multivariate approach to drug product and process development. International Journal of Pharmaceutics, 382(1-2), 23-32. [ Links ]
Hulbert, M., Feely, C., Inman, E., Johnson, A., Kearney, A., Michaels, J., et al. (2008). Risk management in the pharmaceutical product development process. Journal of Pharmaceutical Innovation, 3(4), 227-248. [ Links ]
International conference on harmonization of technical requirements for registration of pharmaceuticals for human use. D (2005). ICH Q9 Quality Risk Management. Geneva, Switzerland. [ Links ]
International conference on harmonization of technical requirements for registration of pharmaceuticals for human use.D (2008). ICH Q10 Pharmaceutical Quality System. PL Geneva, Switzerland. [ Links ]
PC International conference on harmonization of technical requirements for registration of pharmaceuticals for human use.D (2009). ICH Q8R2 Pharmaceutical Development. PL Geneva, Switzerland. [ Links ]
ISO 9004 (2000). Quality management systems - Guidelines for performance improvement. 2.a ed, PL Ginebra, Suiza. [ Links ]
ISO 9000 (2005). Quality management systems - fundamentals and vocabulary. EDN 3.a ed. PL Ginebra, Suiza. [ Links ]
ISO 9001 (2008). Quality management systems - Requirements. EDN 4.a ed, Ginebra, Suiza. [ Links ]
Jadhav, M. y Tambe, S. (2013). Implementation of QbD approach to the analytical method development and validation for the estimation of propafenone hydrochloride in tablet dosage form. Chromatography Research International, 2013, 1-9. [ Links ]
Juran, J. (1992). Juran on quality by design: the new steps for planning quality into goods and services. New York: The Free Press. [ Links ]
Kamm, J. (2007). Can you win the space race? Pharmaceutical manufacturing. [consultado 31 Ago 2014]. Disponible en: http://www.pharmamanufacturing.com/issues/2007/005/ [ Links ]
Lee, Y., Kim, M., Park, M. y Han, K. (2012). Quality by design: understanding the formulation variables and optimization of metformin hydrochloride 750 mg sustained release tablet by Box Behnken design. Journal of Pharmaceutical Investigation, 42(4), 213-220. [ Links ]
Lepore, J. y Spavins, J. (2008). Product quality lifecycle implementation (PQLI) innovations. PQLI design space. Journal of Pharmaceutical Innovation, 3(2008), 79-87. [ Links ]
Lionberger, R., Lee, S., Lee, L., Raw, A. y Yu, L. (2008). Quality by design: concepts for ANDAs. The AAPS journal, 10(2), 268-276. [ Links ]
Maes, I. y Van Liedekerle, B. (2006). The need for a broader perspective if process analytical technology implementation is to be successful in the pharmaceutical sector. Journal of Pharmaceutical Innovation, 1(1), 19-21. [ Links ]
Martin, E., Montague, G. y Robbins, P. (2013). A quality by design approach to process plant cleaning. Chemical Engineering Research and Design, 91(6), 1095-1105. [ Links ]
McGregor, J. y Bruwer, M. (2008). A framework for the development of design and control spaces. Journal of Pharmaceutical Innovation, 3(1), 15-22. [ Links ]
McCurdy, V., Ende, M., Busch, F., Mustakis, J., Rose, P. y Berry, M. R. (2010). Quality by design using an integrated active pharmaceutical ingredient - drug product approach to development. Pharmaceutical engineering, 30(4), 12-33. [ Links ]
Mhatre, R. y Rathore, A. (2009). Quality by design: an overview of the basic concepts. In Quality by design for biopharmaceuticals (pp. 1-8). Hoboken, NJ: John Wiley & Sons. [ Links ]
Naelapää, K., Veski, P. y Bertelsen, P. (2010). Building quality into a coating process. Pharmaceutical Development and Technology, 15(1), 35-45. [ Links ]
Nasr, M. (2006). Risk-based CMC review and quality assessment: what is Quality by Design (QbD). Conferencia FDA/Industria. Philadelphia, PA: School of pharmacy - Temple University. [ Links ]
Noble, P. (2012). Applying Fault Tree Analysis (FTA) as a top level risk management tool in software development. Pharmaceutical engineering, 32(1), 74-81. [ Links ]
Nonaka, I. (1994). A dynamic theory of organizational knowledge creation. Organization Science, 5(1), 14-37. [ Links ]
Nosal, R. y Schultz, T. (2008). PQLI Definition of criticality. Journal of Pharmaceutical Innovation, 3(2), 69-78. [ Links ]
Organización Mundial de la Salud (1996). WHO Expert committee on Specifications for pharmaceutical Preparations - Reporte 34: Anexo 6. Ginebra, Suiza. [ Links ]
Organización Mundial de la Salud (2012). WHO Expert committee on Specifications for pharmaceutical Preparations - Reporte 46: Anexos 3 y 5. Ginebra, Suiza. [ Links ]
Organización Mundial de la Salud (2013). WHO Expert committee on Specifications for pharmaceutical Preparations - Reporte 47: Anexo 2. Ginebra, Suiza. [ Links ]
Politis, S. y Rekkas, D. (2011). The evolution of the manufacturing science and the pharmaceutical industry. Pharmaceutical research, 28(7), 1779-1781. [ Links ]
Portillo, P., Ierapetritou, M., Tomassone, S., Mc Dade, C., Clancy, D., Avontuur, P., et al. (2008). Quality by design methodology for development and scale-up of batch mixing processes. Journal of Pharmaceutical Innovation, 3(4), 258-270. [ Links ]
Potter, C. y Berridge, J. (2010). PQLI Roadmap: Product design, development, and realization, a science- and risk-based approach to implementation - An overview of ISPEs first PQLI guide. Pharmaceutical engineering, 30(2), 1-6. [ Links ]
Prpich, A., Ende, M., Katzschner, T., Lubczyk, V., Weyhers, H. y Bernhard, G. (2010). Drug product modeling predictions for scale-up of tablet film coating - A quality by design approach. Computers and Chemical Engineering, 34(7), 1092-1097. [ Links ]
Rathore, A. (2009). Roadmap for implementation of quality by design (QbD) for biotechnology products. Trends in biotechnology, 27(9), 546-553. [ Links ]
Rathore, A., Bansal, A. y Jaspinder, H. (2013). Knowledge management and process monitoring of pharmaceutical processes in the quality by design paradigm. Advances in Biochemical Engineering/Biotechnology, 132, 217-247. [ Links ]
Rattan, A. y Rubacha, M. (2014). Applying a consistent, compliant and practical riskbased validation process for laboratory systems. Pharmaceutical engineering, 34(2), 1-7. [ Links ]
Reklaitis, G., Khinast, J. y Muzzio, F. (2010). Pharmaceutical engineering science. New approaches to pharmaceutical development and manufacturing. Chemical Engineering Science, 65(21), 4-7. [ Links ]
Riley, B. y Li, X. (2011). Quality by design and process analytical technology for sterile products -where are we now? AAPS PharmSciTech, 12(1), 114-118. [ Links ]
Ring, D., Oliveira, J. y Crean, A. (2011). Evaluation of the influence of granulation processing parameters on the granule properties and dissolution characteristics of a modified release drug. Advanced Powder Technology, 22(2), 245-252. [ Links ]
Rocha, H., Cuadro, J. y Mora, C. (2013). Aplicación de la calidad basada en el diseño (QbD) en la reformulación de tabletas masticables. Revista Colombiana de Ciencias Químico Farmacéuticas, 42(2), 190-214. [ Links ]
Roy, S. (2012). Quality by design: A holistic concept of building quality in pharmaceuticals. International Journal of Pharmaceutical and Biomedical Research, 3(2), 100-108. [ Links ]
Savic, I., Marinkovic, V., Tasic, L. y Krajnovic, D. (2012). From experimental design to quality by design in pharmaceutical legislation. Accredit Qual Assur, 17(6), 627-633. [ Links ]
Shivhare, M. y Mccreath, G. (2010). Practical Considerations for DoE Implementation in Quality by Design. Bioprocess International [consultado 31 Ago 2014]. Disponible en: http://www.bioprocessintl.com/wp-content/uploads/bpi-content/BPI_A_100806AR03_O_98037a.pdf [ Links ]
Somma, R. (2007). Development knowledge can increase manufacturing capability and facilitate quality by design. Journal of Pharmaceutical Innovation, 2(3-4), 87-92. [ Links ]
Staples, M. (2010). The concept of Quality by design. Pharmaceutical Stability Testing to Support Global Markets (pp. 101-106). New York, NY: Springer. [ Links ]
Torbeck, L. y Branning, R. (2009). QbD: Convincing the Skeptics. BioPharm International, 22(5), 52-58. [ Links ]
Trivedi, B. (2012). Quality by design (qbd) in pharmaceuticals. International Journal of Pharmacy and Pharmaceutical Sciences, 4(1), 17-29. [ Links ]
Troup, G. y Georgakis, C. (2013). Process systems engineering tools in the pharmaceutical industry. Computers & Chemical Engineering, 51, 157-171. [ Links ]
Vallejo, B., Cortés, C., Espinosa, A. y Barbosa, H. (2004). Aplicación de la metodología del diseño axiomático en el desarrollo de productos de liberación modificada. Revista Ingeniería e Investigación, 24(3), 41-48. [ Links ]
Van Leeuwen, J., Nauta, M., de Kaste, D., Odekerken-Rombouts, Y., Oldenhof, M., Vredenbregt, M., et al. (2009). Risk analysis by FMEA as an element of analytical validation. Journal of Pharmaceutical and Biomedical Analysis, 50, 1085-1087. [ Links ]
Verma, S., Lan, Y., Gokhale, R. y Burgess, D. (2009). Quality by design approach to understand the process of nanosuspension preparation. International Journal of Pharmaceutics, 377(1-2), 185-198. [ Links ]
Weinberg, S. (2011). Quality by design, in cost-contained regulatory compliance: For the pharmaceutical, biologics, and medical device industries. Hoboken, NJ: Wiley & Sons. [ Links ]
Woodcock, J. (2005). AAPS Workshop - Pharmaceutical quality assessment - a science and risk-based CMC approach in the 21st century. Pharmaceutical quality in the 21st century - an integrated systems Approach. North Bethesda, MD, Octubre 5 -7, 2005. [ Links ]
Wu, H., Khan, M. y Hussain, A. (2007). Process Control Perspective for Process Analytical Technology: Integration of Chemical Engineering Practice Into Semiconductor and Pharmaceutical Industries. Chemical Engineering Communications, 194(6), 760-779. [ Links ]
Wu, H., Tawakkul, M., White, M. y Khan, M. (2009). Quality-by-Design (QbD): An integrated multivariate approach for the component quantification in powder blends. International Journal of Pharmaceutics, 372(1-2), 39-48. [ Links ]
Xu, X., Khan, M. y Burgess, D. (2011). A quality by design (QbD) case study on liposomes containing hydrophilic API: I. Formulation, processing design and risk assessment. International Journal of Pharmaceutics, 419(1-2), 52-59. [ Links ]
Xu, X., Khan, M. y Burgess, D. (2012). A quality by design (QbD) case study on liposomes containing hydrophilic API: II. Screening of critical variables, and establishment of design space at laboratory scale. International Journal of Pharmaceutics, 423(2), 543-553. [ Links ]
Yu, L. (2008). Pharmaceutical quality by design: product and process development, understanding, and control. Pharmaceutical research, 25(4), 781-791. [ Links ]
Zhang, L., Yan, B., Gong, X., Yu, L. y Qu, H. (2013). Application of quality by design to the process development of botanical drug products: a case study. AAPS PharmSciTech, 14(1), 277-286. [ Links ]
