










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Colombiana de Cancerología
Print version ISSN 0123-9015
rev.colomb.cancerol. vol.18 no.3 Bogotá July/Sept. 2014
https://doi.org/10.1016/j.rcc.2014.05.002
REPORTE DE CASO
http://dx.doi.org/10.1016/j.rcc.2014.05.002
Enfermedad de Erdheim-Chester: fractura patológica de húmero izquierdo
Erdheim-Chester Disease: Pathological fracture of the left humerus
Luis Gómeza,b, Camilo Sotoa,b,*, Felipe Criolloa,b, Óscar Messac, Ricardo Romod e Irene Patróne
a Grupo Ortopedia Oncológica, Instituto Nacional de Cancerología, Bogotá D.C., Colombia
b Universidad Militar Nueva Granada, Bogotá D.C., Colombia
c Grupo de Patología Oncológica, Instituto Nacional de Cancerología, Bogotá D.C., Colombia
d Posgrado de Ortopedia, Fundación Universitaria San José, Bogotá D.C., Colombia
e Posgrado de Ortopedia, Pontifícia Universidad Javeriana, Bogotá D.C., Colombia
* Autor para correspondencia.
Correo electrónico: csoto@cancer.gov.co (C. Soto).
Recibido el 27 de febrero de 2014; aceptado el 12 de mayo de 2014
Resumen
La Enfermedad de Erdheim-Chester es una forma rara de histiocitosis que no es de células de Langerhans. Es de etiología desconocida y compromete comúnmente a adultos. Las manifestaciones clínicas son heterogéneas debido a la infiltración histiocítica en múltiples sistemas. Cuando compromete al esqueleto produce dolor óseo, asociado a infiltrados pulmonares intersticiales, cardiacos, hepáticos o renales1-3. Describimos un caso que comienza con dolor óseo crónico y fractura patológica de húmero izquierdo con compromiso poliostótico, sin evidencia de enfermedad multisistémica.
Palabras clave: Erdheim Chester; Fractura; Patológica; Histiocitosis.
Abstract
The Erdheim Chester disease is a rare presentation of non Langerhans cell histiocytosis. The etiology is unknown, and is more common in adults. The clinical manifestations are heterogeneous due to histiocytic infiltration of multiple systems. When there is skeletal involvement, it produces bone pain, and may be associated with interstitial, heart, liver, kidney or pulmonary infiltrates1-3. A case is presented on a patient who began with chronic bone pain and a pathological fracture of the left humerus with polyostotic involvement and with no evidence of multisystemic disease.
Keywords: Erdheim Chester; Fracture; Pathological; Histiocytosis.
Introducción
La enfermedad de Erdheim Chester es una forma rara de histiocitosis donde no están involucradas las células de Langerhans. La edad de presentación está entre la quinta y séptima décadas de la vida, con igual afectación hombre - mujer. La sintomatología se presenta debido a la infiltración de células espumosas a órganos críticos. Los sistemas comúnmente involucrados son: esquelético, nervioso central, renal, pulmonar y piel. Cuando se presenta en el sistema esquelético su manifestación característica es el dolor con áreas de captación en la gammagrafía ósea con Tc-99. Debido a su rareza, es de difícil diagnóstico. En este caso la presentación clínica se confundió con una enfermedad metastásica, y es bajo el enfoque de manejo de ''primario desconocido'' que se dirigió el diagnóstico1-4; con la biopsia ósea y sus estudios complementarios de inmunohistoquímica de dos lesiones independientes, adicionalmente con imágenes y estudios de laboratorio clínico descartamos compromiso de otros órganos, confirmaron el diagnóstico de Enfermedad de Erdheim Chester.
Caso clínico
Se trata de un paciente masculino de 63 años de edad quien acude al servicio de urgencias por presentar dolor y deformidad de su brazo izquierdo luego de realizar un movimiento de tracción y contratracción simple. Al examen físico se evidencia la deformidad de la extremidad, sin compromiso neurovascular. El resto del examen físico fue normal. Las radiografías del área afectada mostraron una fractura de trazo simple en el tercio proximal de la diáfisis del húmero izquierdo, con áreas de osteoesclerosis en un patrón apolillado a lo largo del eje de la diáfisis del hueso (fig. 1a), por lo cual se empezó el estudio de primario desconocido debido a un cuadro de fractura patológica.
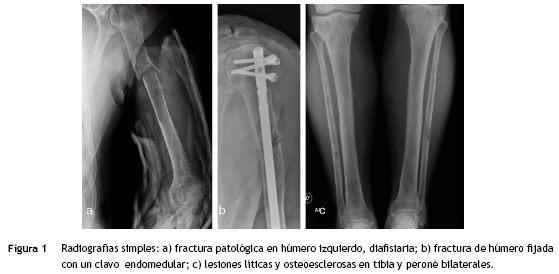
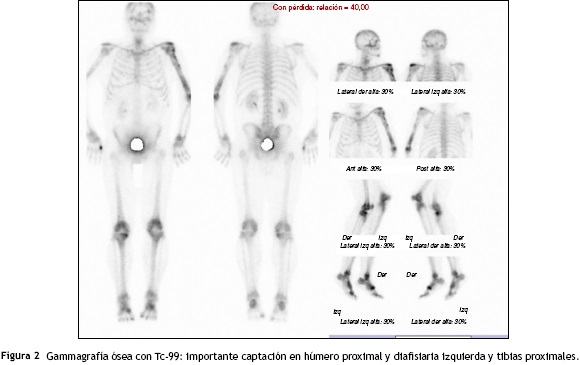
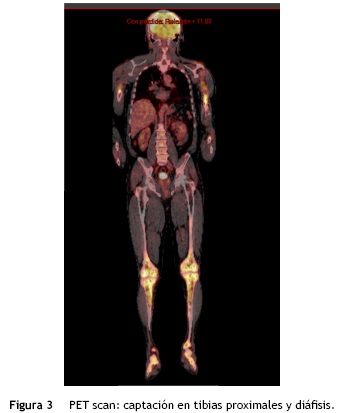
Se solicitó exámenes de laboratorio que resultaron dentro de la normalidad. No obstante, en las radiografías de huesos largos, especialmente en tibia y peroné, se encontró el mismo patrón de lesión esclerosa y lítica saltatoria distribuido en la diáfisis y metafisis de ambos huesos, con compromiso bilateral simétrico (fig. 1c). La gammagrafía ósea mostró hipercaptación metafisiaria y diafisiaria bilateral de tibias (fig. 2). El Pet-Scan reveló los mismos hallazgos de la gammagrafía ósea (fig. 3). Las tomografías de tórax, abdomen y pelvis mostraron ser normales.
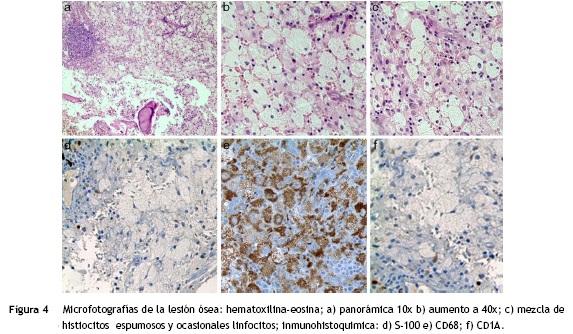
El paciente fue llevado a cirugía de fijación con clavo endomedular de húmero (fig. 1b). El material obtenido del curetaje óseo durante la cirugía fue enviado a patología, la cual reportó: proliferación fusocelular e histiocítica, componente fusocelular hipocelular sin atipia, mitosis ni necrosis; componente histiocítico compuesto por células espumosas grandes de núcleos centrales sin atipia, esta población es débil positiva para CD68 y negativa para S-100 y CD1A. Las coloraciones especiales para microorganismos: Ziehl-Neelsen, Periodic acid-Schiff, Gomory y mucicarmín fueron negativas. Inicialmente existió duda diagnóstica, por lo cual fue necesario realizar biopsia de las lesiones en tibia proximal, cuyo análisis resultó similar al descrito, confirmando el diagnóstico. Se investigó la mutación BRAF-V600E, mediante técnica de PCR utilizando el kit para el equipo Cobas 4800, siendo este resultado negativo. (fig. 4)
Discusión
Existen dos entidades de acumulación de histiocitos espumosos no Langerhans y son la enfermedad de Erdheim Chester y el Xantogranuloma juvenil.
Inicialmente se describió como Granulomatosis Lipídica en 1930 por Jakob Erdheim y William Chester. Esta enfermedad se presenta entre la quinta a séptima décadas de la vida, pero han existido casos de diagnósticos en edades extremas como los 7 años y 82 años de edad. La sintomatología no es específica debido a una variedad de síntomas generales (fiebre, debilidad, pérdida de peso, sudoraciones nocturnas, dolor y en casos pediátricos con alteraciones del desarrollo) y algunas manifestaciones sistémicas como: diabetes insípida, xantelasma, enfermedad pulmonar intersticial y fibrosis retroperitoneal.
La supervivencia a los 5 años es del 68%. Se ha relacionado con una gran variedad de procesos fisiológicos y diversas manifestaciones clínicas que pueden variar desde cursos clínicos indolentes hasta gran compromiso con fallas multiorgánicas fatales que parten de la infiltración de histiocitos espumosos (cargados de lípidos) con citoplasma claro y/o eosinofílico y carentes de gránulos de Birbeck.
Los diagnósticos diferenciales de esta entidad son: osteomielitis, enfermedad de Paget, linfoma, sarcoidosis, metástasis óseas, otras histiocitosis, enfermedad de Gaucher y Niemann- Pick. Todas estas entidades deben descartarse con la clínica de cada una, antes de pensar en una enfermedad de Erdheim Chester2.
Como regla general cuando un paciente se presenta con lesiones óseas sospechosas se debe responder a las siguientes preguntas:
- 1. ¿La anormalidad representa un tumor u otra característica no tumoral? (por ejemplo: infecciones, condiciones inflamatorias, enfermedades metabólicas)
- Si es tumoral, ¿es maligno o es benigno?
- Si es maligno, ¿es de alto grado o de bajo grado?
Todos los pacientes deben ser evaluados con un diagnóstico inicial presuntivo que se enfoque en la historia del paciente. Las características radiográficas de la anormalidad, la edad del paciente, particularmente los pacientes mayores de 40 años tienen mayor riesgo de presentar una enfermedad metastásica, mieloma, linfoma, entre otras; ese escenario es referido como: ''primario desconocido''. Estos pacientes deben ser evaluados con tomografías computarizadas del tórax, abdomen y pelvis, y gammagrafía ósea con Tc-99.
Los exámenes serológicos para antígenos tumorales se realizan para el rastreo de lesiones desconocidas. Los pacientes que inician con una fractura patológica deben tener caracterizada su lesión mediante una biopsia, para asegurar que no se trata de una fractura debida a un sarcoma primario óseo4. Bajo esta perspectiva se enfocó el manejo de este paciente, quien resultó comenzar con una fractura patológica en húmero, cuyo patrón de lesiones poliostóticas incluían lesiones con osteoesclerosis y áreas líticas distales a las rodillas y simétricas bilateralmente. El inicio de la enfermedad de Erdheim Chester con fractura patológica se ha descrito en escasos reportes, apenas uno en tibia y cuello de fémur, lo cual es aún más raro en cuanto a la presentación clínica1,5,6.
El compromiso esquelético ocurre en el 96% de los casos y se manifiesta predominantemente en el miembro inferior con dolor óseo en rodillas, tobillos, tibia, peroné, fémur, y es menos común en cúbito, radio y húmero7. La osteosclerosis es bilateral y simétrica en las diáfisis y en las metáfisis, en regiones epifisiarias y axiales son generalmente escasas y puede inclusive encontrarse periostitis. Se dice que, por lo común, las lesiones son de osteoesclerosis pero pueden hallarse áreas líticas o mixtas en un 5%-30% (particularidad de este caso) e inclusive en huesos planos como costillas y cráneo, lo cual dificulta más el diagnóstico, ya que dichas lesiones líticas suelen encontrarse más frecuentes en histiocitosis de células de Langerhans. Las manifestaciones esqueléticas no suelen ser las únicas al momento del diagnóstico, y se debe buscar siempre la existencia de compromiso en algún otro sistema8.
Radiográficamente se observaron áreas de osteoesclerosis bilaterales y simétricas con lesiones escleróticas3, líticas o mixtas que se semejaban a quistes óseos9. Además de captaciones de Tc-99 en áreas distales, se observaron captaciones proximales en ambas tibias y diafisarias de peroné.
Los criterios diagnósticos de la enfermedad de Erd-heim Chester se basan en: una radiología inespecífica; una gammagrafía que capta el medio de contraste (Tc-99) simétricamente; una histopatología con inmunohistoquímica positiva para CD68+, negativa para CD1a y S1009, y la verificación de ausencia de gránulos de Birbeck, por microscopía electrónica de ser posible. Los hallazgos de laboratorio suelen ser inespecíficos, también como la elevación de la velocidad de eritrosedimentación, el incremento de fosfatasa alcalina, el incremento de la proteína c reactiva3.
La enfermedad de Erdheim Chester es de rara presentación y se caracteriza por la infiltración multiorgánica de histiocitos espumosos no Langerhans. Se cree que esta infiltración se debe a una alteración de la respuesta inmunológica TH1, posiblemente por acción de varias citoquinas, tales como: IL-6, IL-12, IL-4, IL-7, Interferon-a y proteínas quimioatrayentes de monocitos -1 (MCP-1). Todas estas citoquinas juntas subrayan la perturbación inmunológica Th-1 orientada, asociada con esta condición10.
Además, se ha descrito la mutación BRAF-V600E en más de la mitad de los casos publicados2,11. Particularmente, las mutaciones BRAF-V600E se detectaron en alrededor del 54% de los pacientes con enfermedad de Erdheim Chester y en el 38% de las histiocitosis de células de Langerhans, lo que sugiere un origen común12.
Es importante aclarar que no se observó la mutación BRAF-V600E en otros tipos de histiocitosis, como la enfermedad de Rosai Dorfman, con las que puede coexistir12. Recientemente se ha descrito un caso de esta enfermedad con ausencia de mutación BRAF- V600E, pero con la presencia de la mutación NRAS-Q61R. Este hallazgo de mutación oncogénica soporta la hipótesis de que la enfermedad es manejada por la activación de la vía Ras/Raf/MEK/ERK. Lo que hace importante incluir esta mutación en el tamizaje para garantizar la apertura de nuevas oportunidades en terapias dirigidas13. La mutación (BRAF-V600E) se detecta mediante inmunohistoquímica o por PCR sobre las biopsias obtenidas del bloque de parafina14,15.
Los hallazgos de laboratorio pueden mostrarse alterados dependiendo del sistema afectado y el alcance de compromiso en que se encuentre. Debe entonces ampliarse los estudios de laboratorio enfocados al sistema de mayor afectación, se describe por ejemplo: renal, hormonal (pituitaria), pulmonar. Actualmente no se observa un abordaje uniforme de las lesiones o manejo quirúrgico en un tratamiento establecido.
El pronóstico de esta enfermedad empeora si existe compromiso multisistémico, especialmente si se encuentran lesiones cardiovasculares o del sistema nervioso central, no cuando solamente se compromete el esqueleto, como en este caso16. Además el seguimiento del tratamiento de la enfermedad se realiza con PET-scan para conocer el estado de actividad de la enfermedad y para determinar la respuesta a la terapia2.
Se han propuesto diferentes manejos sistémicos como: uso de esteroides, quimioterapia, radioterapia, solos o en combinaciones. De las opciones terapéuticas mencionadas, los corticoides son más recomendados por ser utilizados en un tercio de los estudios. En estudios más recientes se propone el uso de bifosfonatos en pacientes con lesiones de los huesos largos17 con alendronato 70Mg semanales por 9 meses, pamidronato 30mg cada 2 meses por 2 años. Sin embargo, la evidencia no es suficiente para establecer esto como manejo adyuvante18.
Las terapias dirigidas han sido probadas recientemente pero tienen eficacia limitada. El pronóstico en la enfermedad de Erdheim Chester ha mejorado sustancialmente por la terapia con INFα, pero algunas formas refractarias subsisten especialmente en los pacientes con compromiso del sistema nervioso central o cardiovascular.
Se han descrito casos de tratamiento exitoso a dosis terapéuticas iniciales de 3 a 6x106 unidades subcutáneas tres veces por semana, siendo dosis bajas, bien toleradas. Se observa respuesta al tratamiento en aproximadamente un mes, con reducción dramática en los síntomas en pacientes con enfermedad progresiva, y con regresión en las lesiones óseas. El mecanismo de acción no está claro, pero podría deberse a los diversos efectos biológicos de este agente, como: la maduración y activación de células dendríticas; la destrucción de histiocitos mediada por mecanismos inmunológicos (por ejemplo, a través de células asesinas naturales) o por efectos directos antiproliferativos. También hay evidencia anecdótica de terapéutica clínica con beneficios del interferón alfa en otros trastornos histiocíticos (histiocitocis de células de Langerhans y enfermedad Rosai - Dorfaman)19. El tratamiento con interferón fue identificado como predictor independiente de supervivencia20.
El hallazgo de la mutación BRAF-V600E tiene implicaciones significativas en el tratamiento de la enfermedad de Erdheim-Chester. El vemurafenib, un inhibidor del BRAF mutado, fue aprobado recientemente para el tratamiento del melanoma metastásico y en enfermedades con mutaciones BRAF-V600E12 y se describe en pocos casos para pacientes con la enfermedad de Erdheim Chester refractarios al tratamiento con interferón y con esquemas de administración de treinta días a dosis de 1920mg/d y con disminución de la dosis a 960mg/d en el día 30 debido a los efectos colaterales cutáneos. Los resultados documentados en el PET-scan demostraron ser mejores con vemurafenib que los comparados con interferón, aunque la captación ósea es persistente. Pese a esto, los marcadores tumorales y exámenes de imagen demostraron regresión de la enfermedad en aproximadamente 40 días, y más aún al cuarto mes del seguimiento. No se ha determinado el tiempo de duración del efecto terapéutico21.
Reconocimiento
Al Departamento de Biología Molecular del Instituto Nacional de Cancerología por su apoyo en la investigación de la mutación BRAF-V600E, particularmente a Dra. Natalia Olaya, Patóloga-Molecular.
Responsabilidades éticas
Protección de personas y animales. Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datos. Los autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informado. Los
autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de intereses
Los autores declaran que no existe ningún conflicto de interés.
Bibliografía
1. Bindra J, Lam A, Lamba R, Vanness M, Boutin RD. Erdheim-Chester disease: An unusual presentation of an uncommon disease. Skeletal Radiol;. 2013. DOI:10.1007/s00256-013-1793-2. [ Links ]
2. Mazor RD, Manevich-Mazor M, Shoenfeld Y. Erdheim-Chester Disease: a comprehensive review of the literature. Orphanet Journal of Rare Diseases. 2013;8(137):1 -13. [ Links ]
3. Rivera TL, Irish RD, Hoda SA, Steiner GC. Erdheim-Chester Disease: Clinical Pathological Case Discussion. Bulletin of the Hospital for Joint Diseases. 2013;71(2):152-5. [ Links ]
4. Conrad EU. Orthopaedic Oncology: Diagnosis and Treatment: Initial Evaluations of Patients with Musculoskeleta Tumors. Thieme. 2009;1:3-5. [ Links ]
5. Ramos-Font C, Rebollo Aguirre AC, Moral Ruiz A, Bellón Guardia M, Cabello García D, Llamas-Elvira JM. Occult femoral neck fracture in a patient with Erdheim-Chester disease. Rev Esp Med Nucl. 2005;24(6):423. [ Links ]
6. Furutani K, Kurosawa Y, Kageyama T, Kaneko M. A case report of Erdheim-Chester disease. Nihon Igaku Hoshasen Gakkai Zasshi. 1996;56(9):681-3. [ Links ]
7. Haroche J, Arnaud L, Amoura Z. Erdheim-Chester disease. Curr Opin Rheumatol. 2012;24:53 -9. [ Links ]
8. Mazor RD, Kesler A, Shoenfeld Y. Erdheim-Chester Disease: An Orphan Condition Seeking Treatment. IMAJ. 2012;14:388-9. [ Links ]
9. Ota M, Sakamoto M, Sato K. Immunopathological Analysis of Erdheim-Chester Disease with Massive Ascites. Intern Med. 2012;51:2825-30. [ Links ]
10. Arnaud L, Gorochov G, Charlotte F, Lvovschi V, Parizot C. Systemic perturbation of cytokine and chemokine network in Erdheim-Chester disease: a single-center series of 37 patients. Blood. 2011;117(10):2783-90. [ Links ]
11. Lee HJ, Kim TM. Reply to Commentary on ''A Case of Erdheim-Chester Disease with Asymptomatic Renal Involvement''. Cancer Res Treat. 2012;44(4):280. [ Links ]
12. Haroche J, Charlotte F, Arnaud L von Deimling A, Hélias-Rodzewicz Z, Hervier B, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012;120(13):2700-3. [ Links ]
13. Diamond EL, Pentsova E, Borsu L, Chiu AE, Hyman DM, Rosenblum M. Detection of the NRAS Q61R mutation in Erdheim-Chester disease. J Clinic Oncol. 2013;31 15 Supl:1. [ Links ]
14. Blombery P, Wong SQ, Lade S. Erdheim-Chester Disease Harboring the BRAF V600E Mutation. J Clinic Oncol. 2012;30(32):e331-2, doi: 10.1200/JCO.2012.43.2260. [ Links ]
15. Al-Quran S, Reith J, Bradley J, Rimsza L. Erdheim-Chester Disease: Case Report. PCR-Based Analysis of Clonality, and Review of Literature. Mod Pathol. 2002;15(6):666-72. [ Links ]
16. Jaffe R, Julia A, Bridge, Pancras CW, Hogendoorn, Frederik Martens Christopher DM, Fletcher, editores. Erdheim-Chester disease. WHO Classification of Tumors of Soft Tissue and Bone. 4th Ed. Lyon: s.n: IARC; 2013. [ Links ]
17. Asher I, Rabinovith I, Katz M, Sthoeger Z. Erdhiem-Chester Disease in a 49 Year Old Man. IMAJ. 2012;14(6):401. [ Links ]
18. Eyigõr S, Kirazli Y, Memis A, Basdemir G. Erdheim-Chester: The Effect of Bisphosponate treatment-Acase report. Arch Phys Med Rehabil. 2005;86(5):1053-7. [ Links ]
19. Braiteh F, Boxrud C, Esmaeli B, Kurzrock R. Successful treatment of Erdheim-Chester disease, a non- Langerhans-cell histiocytosis, with interferon-α. Blood. 2005;106:2992-4. [ Links ]
20. Arnaud L, Hervier B, Néel A, Hamidou MA, Kahn JE, Wechsler B, et al. CNS involvement and treatment wit interferon-α are independent prognostic factors in Erdheim-Chester disease: a multicenter survival analysis of 53 patients. Blood. 2011;117(10):2778-82. [ Links ]
21. Haroche J, Cohen-Aubart F, Emile JF, Arnaud L, Maksud P, Charlotte F,et al. Dramatic efficacy of vemurafenib in both multisystemic and refractory Erdheim-Cheste disease and Langerhans cell histiocytosis harboring the BRAF V600E mutation. Blood. 2013;121(9):1495-500. [ Links ]
