











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
El mieloma múltiple (MM) es una neoplasia de células B caracterizada por una proliferación descontrolada de células plasmáticas cuyo origen inicial es la médula ósea y puede acompañarse de nichos extramedulares denominados plasmocitomas. Estas células clonales secretarán un tipo específico de inmunoglobulina (habitualmente IgG o IgA) el cual se denomina componente M o monoclonal, detectable en muestras serológicas y de orina; al mismo tiempo, un elemento particular de estas inmunoglobulinas, las cadenas ligeras (kappa o lambda) será detectable con mayor facilidad a nivel urinario y se asocian a un deterioro del funcionamiento renal para el diagnóstico1,2.
La presentación clínica del mieloma múltiple es diversa y va más allá de las manifestaciones clínicas clásicas de dolor lumbar crónico, fracturas localizadas en huesos sometidos a una mayor exigencia mecánica (fémur o húmero), anemia (<10 g/dL), enfermedad renal crónica (creatinina sérica mayor de 2 mg/dL o aclaramiento renal menor de 40 mL/min)3,4.
El objetivo de esta revisión es brindar un panorama general del proceso diagnóstico del mieloma múltiple, así como las diferentes herramientas disponibles para el diagnóstico y referencia oportuna.
Epidemiología en América Latina
El MM constituye el 1% de todas las neoplasias malignas, y hasta un 10% de las neoplasias hematológicas5. Alrededor del mundo, su incidencia es de 4 casos por cada 100,000 habitantes, pero acorde con registros realizados tanto en Estados Unidos como en Europa, la población afroamericana se ve afectada con una mayor frecuencia en comparación con la población asiática o latina6. A pesar de haber sido considerada inicialmente una enfermedad del adulto mayor, cada vez resulta más frecuente su ocurrencia en pacientes jóvenes, acompañada de la expresión de características genéticas de mal pronóstico7. Aunque en la actualidad existen múltiples fármacos con mecanismos novedosos para el tratamiento tanto de pacientes con diagnóstico de novo como en recaída, en América Latina la mortalidad continúa siendo considerable. Curado y colaboradores, tras analizar la epidemiología del MM en 17 países de Latinoamérica, reportaron que la incidencia más alta de la región se encuentra en Cali (Colombia) con 14.2 casos por 100,000 habitantes; respecto a la mortalidad, Guatemala es el país con mayor proporción de muertes (12.5%), y la más baja ocurre en Brasil con el 1.4%; también indican que la edad promedio es de 60 años, sin mostrar alguna preferencia por el género8. En México, el MM representa el 4.2% de todas las neoplasias hematológicas (cifra que dobla la incidencia promedio observada en población caucásica)9, y es la discrasia de células plasmáticas más frecuente con el 65% del total, seguida de la amiloidosis (13%), la macroglobulinemia de Waldestrom (12%) y la gammapatía monoclonal de significado incierto (MGUS, por sus siglas en inglés "Monoclonal gammopathy of undetermined significance") (10%)10.
Manifestaciones clínicas y su relación con el diagnóstico
Históricamente, las manifestaciones clínicas clásicas de los pacientes con MM son: anemia, la hypercalcemia, lesiones óseas y deterioro del funcionamiento renal (CRAB: acrónimo en inglés de los síntomas: calcium, renal, anemia, bone). En la época donde carecíamos de estudios sofisticados, frecuentemente bastaba la sospecha clínica del CRAB para considerar el inicio del tratamiento de soporte (bifosfonatos, eritropoyetina, hemodiálisis) y el tratamiento definitivo (inmunomoduladores, esteroides o quimioterapia)11.
Ahora, seguimos reconociendo la utilidad clínica del CRAB para orientar la sospecha diagnóstica, e incluso continúa siendo de total utilidad para discriminar entre los casos activos y sintomáticos, de aquellos que solo muestran actividad bioquímica en suero y permanecen asintomáticos como el MM asintomático y la gammapatía monoclonal de significado incierto12. Es por ello que, de acuerdo con la última actualización presentada por el Grupo internacional de trabajo del mieloma múltiple (por sus siglas en inglés IMWG: "International Myeloma Working Group"), estas manifestaciones clínicas ahora se encuentran englobadas en lo que se llaman eventos definitorios de mieloma múltiple13.
Dentro de los diferentes biomarcadores que se han adicionado a los eventos definitorios de MM se encuentran: la confirmación de la clonalidad de las células plasmáticas identificadas en el extendido de médula ósea, la presencia de cadenas ligeras libres (FLC) en suero > 100 mg/L o la presencia de una o más lesiones óseas identificadas a través de una resonancia magnética.
A pesar de tener todo este arsenal diagnóstico, alrededor de un 2% de los pacientes con MM no mostrarán evidencia de algún componente monoclonal detectado tanto por la electroforesis como por inmunofijación13. En la Tabla 1 se presentan los criterios diagnósticos de MM al igual que los eventos considerados como definitorios por el IMWG12,13. La Figura 1 resume los principales cuadros clínicos que desarrollan los pacientes con MM y que se detallarán a continuación.
Tabla 1 Criterios diagnósticos del Grupo internacional de trabajo de mieloma múltiple y los trastornos relacionados con las células plasmáticas.
| Trastorno | Definición de la enfermedad |
|---|---|
| Mieloma múltiple latente | Ambos criterios deben cumplirse: • Proteína monoclonal en suero (IgG o IgA) >3 g/dL, o proteína monoclonal urinaria > 500 mg por 24 horas y/o células plasmáticas de médula ósea clonales 10% a 60% • Ausencia de eventos definitorios de mieloma |
| Mieloma Múltiple | Ambos criterios deben cumplirse • Células plasmáticas en médula ósea >10% o plasmocitoma extramedular comprobado por biopsia. • Uno o más de los siguientes eventos que definen mieloma: a. Hipercalcemia: calcio sérico > 11 mg/dL b. Insuficiencia renal: aclaramiento de creatinina <40 ml por minuto por MDRD c. Anemia: valor de hemoglobina <10 g/dL d. Lesiones óseas: una o más lesiones osteolíticas al menos de 5 mm de tamaño en la radiografía, tomografía computarizada o tomografía por emisión de positrones e. Porcentaje de células plasmáticas de médula ósea > 60% f. Relación de cadenas ligeras libres en suero >100 mg/L |
| Gammapatía monoclonal de importancia indeterminada no IgM | Los 3 criterios deben cumplirse: • Proteína monoclonal tipo no IgM en suero <3 g/dL • Infiltración linfoplasmocítica de médula ósea <10% No hay evidencia de daño a órgano blanco |
MDRD: Fórmula abreviada para la detección de insuficiencia renal crónica en atención primaria.
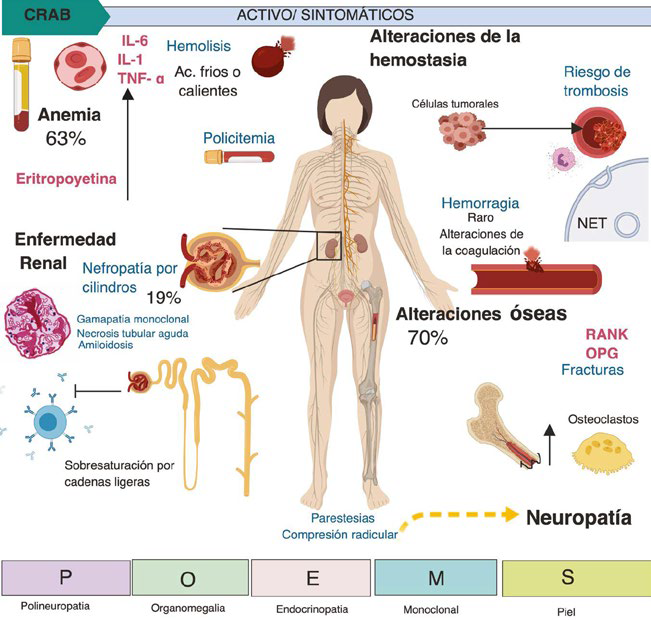
Ac: anticuerpos; CRAB: C: calcio elevado, R: riñón, daño renal, A: anemia, B: bone, lesiones óseas; IL-1: interleucina 1; IL-6: interleucina 6; NET: trampas extracelulares de neutrófilos; OPG: osteoprotegerina; RANK: activador del receptor del factor nuclear kappa beta; TNF-a: factor de necrosis tumoral alfa.
Figura 1 Presentaciones clínicas del mieloma múltiple
Manifestaciones clínicas asociadas al MM Anemia
La anemia es la manifestación más frecuente de MM; alrededor del 60% de los pacientes muestran manifestaciones de síndrome anémico al momento del diagnóstico. La fisiopatología de la anemia en estos casos tiene múltiples vías, siendo la disminución en número de progenitores hemáticos a favor de las clonas neoplásicas en médula ósea la principal condicionante; seguida de una deficiencia de eritropoyetina secundaria a la enfermedad renal concomitante o a las pérdidas hemáticas por sangrados activos u ocultos14. Otros mecanismos relacionadas al descenso de la hemoglobina en estos pacientes son la depleción de las reservas de hierro, y la sobreproducción de diversas interleucinas como IL-6, IL-1 o TNF-alfa2,15. Curiosamente, debido a que dentro de la fisiopatología del MM se encuentran implicadas vías de señalización como lo son la Janus kinasa (JAK2), algunos casos muy aislados pueden mostrar dentro de su cuadro clínico policitemia, particularmente policitemia vera concomitante16. Existen también situaciones poco frecuentes que propician la anemia, como la hemolisis mediada por anticuerpos fríos o calientes, y la mielotoxicidad asociada a los diferentes fármacos implicados en el tratamiento del MM15.
Gascón y colaboradores identificaron, en el registro Europeo de Anemia en Cáncer, que a diferencia de los pacientes con linfoma, los casos con MM mostraron anemia con mayor frecuencia de (85.3%), con cifras de hemoglobina en el rango de los 10-11.9 g/dL de hemoglobina17.
Es altamente relevante identificar y manejar la anemia para evitar las complicaciones cardiovasculares y la disminución en la calidad de vida18. El principal tratamiento de la anemia asociada al MM consistía en la administración de concentrados eritrocitarios, pero debido a las diferentes complicaciones relacionadas con la transfusión, esta práctica se ha abandonado y ha sido sustituida por los diferentes tipos de eritropoyetinas recombinantes19. El uso de estimulantes de la eritropoyesis está considerado dentro de las diferentes estrategias de soporte tanto al momento del diagnóstico como durante la progresión de la enfermedad, a pesar de que su eficacia para incrementar los niveles de hemoglobina sea menor que las transfusiones (60-75% versus 35%)20. Además, en la actualidad las diferentes terapias blanco muestran un incremento considerable en las respuestas hematológicas; por tanto, el uso de las diferentes eritropoyetinas recombinantes se reserva a aquellos pacientes sintomáticos o cuya hemoglobina se mantiene menor a 8 g/dL, y actualmente no hay ensayos que valoren su eficacia en pacientes atendidos con esquemas basados en inmunomoduladores como lenalidomida, inhibidores de proteosoma o los nuevos anticuerpos monoclonales17.
Neuropatía
Durante décadas se consideró a la neuropatía como una manifestación secundaria a la actividad del MM, teniendo como origen la compresión radicular o medular por las lesiones líticas, el depósito de amieloide o la presencia de crioglobulinemia21. Actualmente, con el éxito de los tratamiento oportunos, la neuropatía sigue presente en el espectro clínico del MM, pero debida a los efectos secundarios del uso de ciertos medicamentos (talidomida y bortezomib), y cuya severidad puede complicar el éxito del tratamiento y la calidad de vida de los pacientes22,23. La mayoría de los casos presentan disestesias y parestesias en manos y pies, en menor medida hiperestesia o debilidad muscular, y una minoría de los casos reporta neuropatía autonómica como hipotensión, bradicardia, impotencia y constipación, especialmente en casos con una neuropatía previa concomitante24,25. Otras manifestaciones neurológicas menos frecuentes son el síndrome del túnel de carpo (especialmente en pacientes con amiloidosis primaria o secundaria a MM)26; y dolores articulares, tanto poliarticulares como monoarticulares, muy semejantes a las descritas en artritis reumatoide; al respecto, Eisenman y colaboradores describieron en su serie de 101 pacientes que cerca de un 22% de los casos se identificaron con un daño articular independiente de la infiltración tumoral27.
Anormalidades en hemostasia
En forma semejante a lo que ocurre con otro tipo de tumores, los pacientes con MM presentan un riesgo de trombosis elevado, e incluso la trombosis puede ser la primera manifestación clínica de pacientes con MGUS o MM28. Al respecto, la teoría más plausible hasta ahora señala que las células tumorales son capaces de secretar factores de crecimiento tisular y liberar exosomas que interactúan con los neutrófilos circulantes mediante sus trampas extracelulares denominadas NET (por las siglas neutrophil extracelular traps), ocasionando daño endotelial y favoreciendo así la formación de trombos29,30.
El uso de inmunomoduladores (talidomida, tenalidomida) en el tratamiento del MM también eleva el riesgo de trombosis. Debido a esto, algunos grupos han probado la eficacia de adicionar al tratamiento antiagregantes plaquetarios como ácido acetilsalicílico o heparina de bajo peso para reducir el riesgo trombótico, obteniendo resultados favorables y equiparables (reducción de eventos trombóticos en 2.27% y 1.2% respectivamente)31.
Además del riesgo de trombosis, los pacientes con MM poseen un riesgo elevado de hemorragia. Este riesgo se asocia tanto a una disminución del número de plaquetas circulantes, como a una interacción del componente M sobre el funcionamiento de los factores de coagulación. Alrededor de un tercio de los casos muestran anormalidades por lo menos en un factor de coagulación; siendo el fibrinógeno y el factor VIII los más importantes, clínicamente se puede sospechar de este mecanismo activo en un paciente que presente alteraciones en los valores tanto del tiempo de trombina (TT) como del tiempo parcial de tromboplastina (TTPa) respectivamente32.
El desarrollo de hemofilia secundaria es una manifestación hemostática poco frecuente en MM. Recordemos que la hemofilia es un padecimiento relativamente raro (0.2 -1.9 por millón de habitantes) y de estos, hasta el 10% de los casos se relaciona con algún tipo de neoplasia linfoproliferativa, como los linfomas o el mieloma. Se postula que el desarrollo de la hemofilia se produce por la acción de las inmunoglobulinas sobre el factor VIII, prolongando el TTPa de manera considerable en pacientes con MM e incluso desde etapas tempranas como MGUS33.
Alteraciones óseas
El MM incrementa 9 veces el riesgo de fractura, siendo los principales sitios las vértebras y las costillas, de las cuales hasta un 11% son de tipo incidental34. Teniendo así que alrededor de un 70% de los pacientes con MM mostraran lesiones esqueléticas al diagnóstico y un 85% lo desarrollará posteriormente al mismo, e incluso, en el 20% de aquellos que se fracturen será motivo de muerte35. En los pacientes con MGUS también se incrementa el riesgo de fracturas principalmente a nivel vertebral (riesgo relativo de 2.7), lo que puede sugerir progresión hacia MM38. De especial interés son las lesiones óseas centrales, es decir, aquellas que se presenten en las estructuras del sistema nervioso central (cráneo y columna vertebral), dado que tienen valor pronóstico respecto a una menor supervivencia39. Hoy en día, disponemos de puntajes para valorar la supervivencia de pacientes que presenten metástasis de MM en columna vertebral, como lo son: el Tokuhashi, Sioutos, Tomita, Van der Linden y Bauer, siendo este último el que muestra una mayor asociación con la supervivencia en pacientes con metástasis40. Al aplicar estos puntajes en pacientes con MM sometidos a un procedimiento quirúrgico vertebral, no muestran una correlación con el pronóstico ya que tanto el puntaje de Tomita como el de Tokuhashi subestiman la supervivencia de estos pacientes, sugiriéndose guiar el pronóstico acorde con el Sistema Internacional de Estratificación (ISS)40.
Los avances en la fisiopatología del MM han permitido entender su asociación con otras células en lo que se denomina microambiente. Son diversas las vías de señalización celular implicadas, especialmente las relacionadas con la proliferación celular y la carcinogénesis, como la vía RANK (del inglés: Receptor Activator of Nuclear Factor K B), su ligando RANKL y osteoprotegerina (OPG)36. La relevancia de esta vía radica en que permite la diferenciación de los osteoclastos con su subsecuente activación, rompiendo el balance óseo y generando las diferentes lesiones líticas características del MM, siendo esta una de las actuales líneas de investigación con fines terapéuticos37.
Enfermedad renal
El deterioro del funcionamiento renal es una de las principales causas de complicaciones relacionadas al MM. A pesar de que en su mayoría el deterioro del funcionamiento renal es relacionado al componente M, la mayoría de las lesiones renales se agrupan en la nefropatía por cilindros, necrosis tubular aguda, nefritis tubulointersticial, amiloidosis, gammapatía monoclonal con depósito de inmunoglobulinas, glomerulopatía inmunotactoide, crioglobulinemia tipo I, glomerulonefritis proliferativa con depósito de IgG, glomerulopatía por C3 con gammapatía glomerular y la histiocitosis por depósito de cristales41.
Debido a su tamaño, las inmunglobulinas no pueden filtrarse, pero sí pueden interactuar con las diferentes células del mesangio ocasionando los depósitos observables histopatológicamente; estos depósitos pueden integrarse por cadenas ligeras, ligeras y pesadas o exclusivamente pesadas, siendo la variante de cadenas ligeras la más frecuente (19%)43.
Por otro lado, las cadenas ligeras libres si pueden filtrarse y absorberse en el túbulo contorneado proximal por mecanismos de endocitosis dependiente de clatrinas a través de los receptores de megalina/ cubilina; la sobreproducción de cadenas ligeras libres sobresatura este sistema contribuyendo con diferentes tipos de alteraciones tubulares42.
La alteración renal que se describe con mayor frecuencia es la nefropatía por cilindros compuestos de cadenas ligeras (Kappa o Lambda); en biopsias renales su frecuencia es variable, alcanzando hasta un 47.5%, mientras que la presencia de amiloidosis renal se representa en 7.1 % de los casos44. La uromodulina (proteína de Tamm Horsfall) es fundamental para el desarrollo de la nefropatía por cilindros ya que puede inducir agregados con los diferentes compuestos de cadenas ligeras; posteriormente a esto, se induce la activación de neutrófilos y la consecuente activación de vías de señalización como la del factor nuclear kB45,46. A pesar de que la mayor parte de los complejos son resultado de la interacción con las cadenas ligeras de inmunoglobulinas, recientemente se han descrito casos de cilindros compuestos principalmente de amiloide47.
Debido al excedente de cadenas ligeras, los pacientes pueden excretar cantidades significativas de glucosa, bicarbonato y potasio al igual que aminoácidos, entidad que al acompañarse de osteomalacia integra el síndrome de Fanconi secundario (FS)48. En la Clínica Mayo, en un estudio realizado entre 1968 y 2002, se identificaron 32 casos con FS relacionados en su mayoría con MM (31%), macroglobulinemia de Waldenstrom (6%), mieloma asintomático (19%) y MGUS (44%)49. Otra de las anormalidades descritas solo en casos aislados es la enfermedad de Goodpasture; esta complicación es causada por depósitos de inmunoglobulina que causan hemorragia pulmonar, anemia, neumonitis intersticial y glomerulonefritis50.
Actualmente, se reconoce el recambio plasmático como una estrategia terapéutica eficaz en circunstancias como el depósito de cadenas ligeras, complejos inmunes, auto anticuerpos y crioglobulinas51,52.
Estudios de utilidad para el diagnóstico "¿Qué es esto?"
"¿Qué es esto?", con esta frase concluye la carta redactada por el Dr. William MacIntyre dirigida al Dr. Henry Bence Jones, fechada el 1° de noviembre de 1845, y que trata acerca de una muestra de orina con características anormales en un paciente con sospecha de nefrosis y dolor lumbar incapacitante53; a la muerte del paciente, John Dalrymple ejecutó la necropsia y describió un patrón histológico anormal en la mayoría de los huesos del difunto; 30 años después, Waldeyer describió esa morfología celular y la bautizó como células plasmáticas54. Gracias a estos hallazgos, en conjunto se realizaron las primeras vinculaciones entre los hallazgos clínicos y alteraciones de laboratorio en pacientes con MM y que continúan vigentes hasta la actualidad.
Los estudios útiles para el diagnóstico de MM los podemos dividir en cuatro grandes rubros (Ver Figura 2):
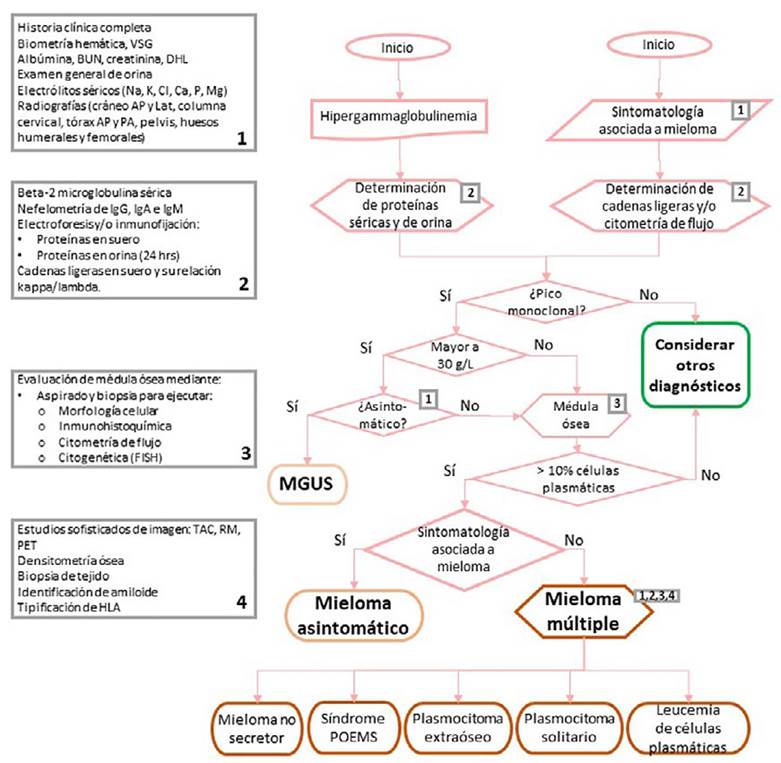
Los rectángulos grises indican los estudios complementarios al diagnóstico: 1) Iniciales o de utilidad para la sospecha, 2) diagnóstico de proteínas, 3) conformación diagnóstica, 4) complementos diagnósticos. Se indica en que parte del proceso serán de utilidad cada uno de los mismos. AP: anteroposterior; BUN: nitrógeno ureico en sangre; Ca: calcio; Cl: cloro; DHL: deshidrogenasa láctica; FISH: hibridación fluorescente in situ; HLA: antígenos leucocitarios humanos; IgA: inmunoglobulina A; IgG: inmunoglobulina G; IgM: inmunoglobulina M; K: potasio; Lat: lateral; Mg: magnesio; MGUS: gammapatía monoclonal de significado incierto; Na: sodio; P: fósforo; PA: posteroanterior; PET: tomografía por emisión de positrones; POEMS: síndrome de polineuropatía, organomegalia, endocrinopatía, componente monoclonal y piel; RM: resonancia magnética; TAC: tomografía computarizada; VSG: velocidad de sedimentación glomerular.
Figura 2 . Algoritmo diagnóstico de los desórdenes de células plasmáticas
Iniciales o útiles para la sospecha
Es fundamental partir de la elaboración de una historia clínica completa, poniendo especial énfasis en las manifestaciones clínicas previamente descritas; partiendo de ahí, se solicitarán estudios paraclínicos disponibles en casi cualquier unidad médica: biometría hemática con frotis (búsqueda de apilamiento anormal de los eritrocitos, fenómeno denominado Rouleaux); química sanguínea, incluyendo proteínas séricas, electrolitos séricos, radiografías simples. Es importante recordar que algunos pacientes con MM, y particularmente MGUS, son asintomáticos y el abordaje comienza al detectarse en alteraciones en los paraclínicos de rutina55,56.
Diagnóstico de proteínas
El siguiente paso ante la sospecha de MM es evidenciar la presencia del componente M en sangre u orina (preferentemente en ambas) mediante la cuantificación de inmunoglobulinas séricas, electroforesis de proteínas (en orina, suero o ambos), electroforesis por inmunofijación (en suero, orina o ambos), y proteínas totales en orina de 24 horas. Cuando la electroforesis de proteínas séricas resulta negativa está indicada la inmunofijación y prueba de cadenas ligeras. En este paso también debe aprovecharse la oportunidad para determinar la beta-2-microglobulina sérica, a fin de poder calcular el ISS para establecer el pronóstico del paciente13,57.
Con los elementos recabados hasta este punto tendremos la certeza para descartar o reorientar el proceso diagnóstico hacia otras patologías, como infecciones virales (hepatitis C, virus de la inmunodeficiencia humana), enfermedades autoinmunes o hepatopatías; o bien de afirmar la presencia de algún desorden de células plasmáticas; si el paciente permanece asintomático nos orientará hacia MGUS, en caso de síntomas, debemos continuar el proceso diagnóstico de MM7,56.
Confirmación diagnóstica
El paso ineludible en el diagnóstico de MM es la evaluación de médula ósea mediante aspirado y/o biopsia, a fin de comprobar la presencia anormal de clonas plasmáticas, así como el porcentaje y patrón de infiltración a médula ósea. Resulta indispensable determinar el origen clonal neoplásico de las células observadas, por lo que además de la valoración morfológica con tinciones habituales deberán realizarse tinciones inmunohistoquímicas, y la valoración del fenotipo por citometría de flujo (CD38, CD138, CD79a, CD56, CD117, CD20, CD52 y CD10)1,57.
En la era de las medicinas con blancos ultra específicos, es necesario también realizar el cariotipo e hibridación fluorescente in situ (FISH, del inglés fluorescence in situ hybridization) para conocer el perfil de riesgo genético con fines pronósticos, y de ser posible, elegir fármacos específicos. A propósito de estas pruebas, recordar que el riesgo genético en MM se divide en 2 grupos de acuerdo con las alteraciones presentes: riesgo alto: t(4;14), t(14;16), t(14;20), deleción de 17p (mediante FISH), deleción del cromosoma 13 e hipodiploidia; riesgo estándar: ausencia de cualquier alteración de alto riesgo, hiperdiploidía, t(11;14) y t(6;14) (mediante FISH)1,56,57.
Complementos diagnósticos
Bien sea en la etapa de sospecha diagnóstica o una vez obtenida la confirmación diagnóstica de MM, es indispensable efectuar estudios complementarios para conocer la totalidad del espectro clínico de la enfermedad en el paciente. En primer lugar, tendremos los estudios de imagenología como la resonancia magnética, tomografía computarizada o por emisión de positrones, así como la densitometría ósea, que permitirán la evaluación más detallada de las manifestaciones óseas del MM, especialmente útil en subtipos particulares como lo es el plasmocitoma solitario o extraóseo.
También será de utilidad ejecutar la tipificación de HLA y determinación de amiloide en casos donde se tenga contemplado efectuar un trasplante de células progenitoras hematopoyéticas, o bien, confirmar la presencia de amiloidosis13,57.
Consideraciones finales
Como en todo abordaje diagnóstico, ante la sospecha de MM la directriz principal del abordaje es el cuadro clínico del paciente, aunque no debe despreciarse la proporción de casos que son detectados oportunamente por alteraciones en los laboratorios sin alguna otra causa aparente. Con el algoritmo diagnóstico presentado también deseamos comunicar que este proceso no es labor exclusiva del especialista hematólogo; como mencionamos en el texto, casi todos los estudios de apoyo diagnóstico en los primeros niveles del algoritmo se encuentran disponibles en la mayoría de unidades médicas, incluso en centro de atención primaria; así entonces, se espera que conforme se avanza en el algoritmo el paciente sea referido a niveles superiores de atención médica para encausar mejor el protocolo diagnóstico usando las técnicas más modernas y sofisticadas, las cuales enteramente recomendamos sean indicadas por el especialista hematólogo a fin de optimizar los recursos.
En conclusión, el mieloma múltiple es una enfermedad con eventos clínicos bien definidos que llevan al diagnóstico o que son parte de la historia de la enfermedad, ya sea por progresión o ante las terapéuticas aplicadas. El médico debe tener conocimientos actualizados sobre los mecanismos moleculares subyacentes en todo momento para prestar atención a las presentaciones atípicas del MM, siendo estas cada vez más frecuentes, todo ello con la finalidad de instaurar oportunamente o modificar las estrategias terapéuticas para disminuir la morbilidad y mantener la mayor calidad de vida posible para el paciente

