










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkTecnura
Print version ISSN 0123-921X
Tecnura vol.18 no.spe Bogotá Dec. 2014
https://doi.org/10.14483/udistrital.jour.tecnura.2014.DSE1.a08
DOI: http://doi.org/10.14483/udistrital.jour.tecnura.2014.DSE1.a08
Automation of functional annotation of genomes and transcriptomes
Automatización de la anotación funcional de genomas y transcriptomas
Luis Fernando Cadavid Gutiérrez*, José Nelson Pérez Castillo**, Cristian Alejandro Rojas Quintero***, Nelson Enrique Vera Parra****
* Medicine Doctor, Ecology and Evolutionary Biology PhD., IEI Research Group - Teacher / Researcher, Institute of Genetics and Department of Biology, National University, Bogotá D.C., Colombia. lfcadavidg@unal.edu.co
** System Engineer, Informatics PhD., GICOGE Research Group - Director of Center for Scientific Research and Development, Universidad Distrital Francisco José de Caldas, Bogotá D.C., Colombia. nelsonp@udistrital.edu.co
*** System Engineer Student, GICOGE Research Group - Student, Universidad Distrital Francisco José de Caldas, Bogotá D.C., Colombia. carojasq@correo.udistrital.edu.co
**** Electronic Engineer, Information Sciences and Communication M.Sc., GICOGE Research Group - Teacher / Researcher, Universidad Distrital Francisco José de Caldas, Bogotá D.C., Colombia. neverap@udistrital.edu.co
Fecha de recepción: June 10th, 2014 Fecha de aceptación: November 4th, 2014
Citation / Para citar este artículo: Cadavid Gutiérrez, L. F., Pérez Castillo, N. J., Rojas Quintero, C. A., & Vera Parra, N. E. (2014). Automation of functional annotation of genomes and transcriptomes. Revista Tecnura, 18 (Edición especial doctorado), 90-96. doi: 10.14483/udistrital.jour.tecnura.2014.DSE1.a08
Abstract
Functional annotation represents a way to investigate and classify genes and transcripts according to their function within a given organism.
This paper presents Massive Automatic Functional Annotation (MAFA - Web), which is an online free bioinformatics tool that allows automation, unification and optimization of functional annotation processes when dealing with large volumes of sequences. MAFA includes tools for categorization and statistical analysis of associations between sequences. We have evaluated the performance of MAFA with a set of data taken from Diploria-Strigosa transcriptome (using an 8-core computer, namely E7450 @ 2,40GHZ with 256GB RAM), processing rates of 2,7 seconds per sequence (using Uniprot database) and 50,0 seconds per sequence (using Non-redundant from NCBI database) were found together with particular RAM usage patterns that depend on the database being processed (1GB for Uniprot database and 9GB for Non-redundant database). Aviability: https://github.com/BioinfUD/MAFA.
Keywords: Annotator, Functional annotation, Gene ontology, High Throughput Sequencing.
Resumen
La anotación funcional es un medio para investigar y clasificar genes y transcritos de acuerdo con la función que realizan en un organismo dado.
Este artículo presenta Massive Automatic Functional Annotation (MAFA - Web), la cual es una herramienta bioinformática libre y en línea que permite la automatización, unificación y automatización de los procesos de la anotación funcional, trabajando con grandes volúmenes de secuencias. MAFA incluye herramientas para la categorización y análisis estadístico de las asociaciones entre secuencias y su ontología correspondiente. Se ha evaluado el desempeño de MAFA con un set de datos tomado del transcriptoma de Diploria-Strigosa (usando un computador de 8 núcleos, específicamente un E7450 @ 2,40GHZ con 256GB de memoria RAM). Se encontraron tasas de procesamiento de 2,7 segundos por secuencia (usando la base de datos de Uniprot) y 50,0 segundos por secuencia (usando la base de datos Non-redundant de NCBI), junto con un patrón particular de uso de RAM que depende de la base de datos que es procesada (1GB para la base de datos Uniprot y 9GB para la base de datos Non-redundant). Disponibilidad: https://github.com/BioinfUD/MAFA.
Palabras clave: anotador, anotación funcional, ontología génica, secuenciación de alto rendimiento.
Introduction
Biological-sequence decoding plays an essential role in almost all research branches of Biology. For various decades, sequencing processes were conducted using the Sanger method (including the human genome project, where this method was crucial). However, the cost of the method and its limitations in terms of performance, scalability, speed and resolution have led to a migration trend towards using new procedures in the last 5 years, namely the so called "next generation sequencing" (Mekster, 2010; Martin & Wang, 2011) These new technologies allow having lower-cost, more-efficient sequencing, which leads to an exponential growth in the volumes of sequenced data.
Optimization of the sequencing process would be worthless without the development and optimization of suitable computing tools capable of analyzing such large sequenced-data volumes. In this context, one of the main needs of genomic-transcriptomic data mining is functional annotation. As a process, functional annotation consists of two stages, namely a search for known similar sequences (through alignment) and the association of such sequences to functional categories. The type of tools that are commonly used to carry out functional annotation processes are the following: BLAST - Basic Local Alignment Search Tool (Altschul et al., 1990), (Camacho et al., 2008) (for finding sequences through alignment) and GO - Gene Ontology (Ashburner et al., 2000) (which provides controlled-term vocabulary to describe particular genes and the gene-product attributes within a particular organism).
The annotation process for unknown sequences involves the use and integration of various tools that deal with the following tasks: Local-alignment search for comparing unknown sequences with known-sequence databases (e.g. Swissprot, Uniprot, Refseq, among others), association between sequences and the ontology that describes the functionality of such sequences and categorization and statistical analysis of the corresponding associations).
This paper is divided in two sections. In the first section we describe the software working way. In the second section we have made an evaluation of MAFA using various datasets.
Methodology
General description
MAFA is a free online bioinformatics tool that has been optimized to carry out functional annotation processes over large numbers of nucleotide sequences (genomes and transcriptomes). Moreover, MAFA includes additional tools to perform categorization and statistical analysis of the corresponding sequence-ontology associations. MAFA is intended to operate by a web interface making the functional annotation a simple process (almost intuitive) for biologist.
Architecture
MAFA consists of 4 modules that constitute a work flow. In order to run and integrate the modules, it is necessary to use additional tools that apply to all modules. Figure 1 shows the 4 modules together with the work flow and the cross-module applicable tools.
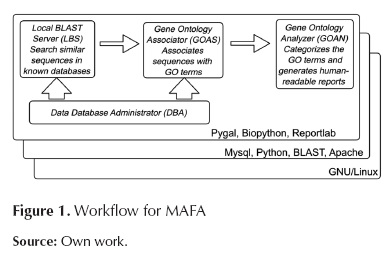
Cross-module software components
MySQL: A relational and multi-thread, multi-user data-base management system, also free software. GNU/Linux: A free operating system that is suitable for servers and also for running bio-informatics tools. Biopython (Cock et al., 2009): has proposed this free software project with various modules intended to facilitate manipulation of bioinformatics data. Pygal: Free libraries that assist the production of graphical materials for the representation of information. BLAST (Basic Search Alignment Tool): A tool intended to find local regions of similarity through sequence alignment. Reportlab: A open-source Pyton-based library that facilitates the creation of PDF-format files. Apache: A HTTP server with free license.
Local BLAST server
This module is in charge of running BLAST (Nucleotides vs Amino-acids) and also of storing the corresponding output using the XML format. Figure 2 shows the inputs and outputs of this module. The script involved in this module is as follows:
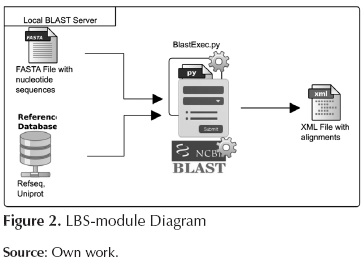
BlastExec.py: This script orders the system to run blastx (Sequences against Reference database) using various cores.
GO associator
This module establishes the existing associations between the best hits, obtained from BLAST, and the terms from Gene Ontology. These associations are made by means of mapping tables between sequence identifiers and GO terms. The GOAS module is shown in Figure 3.
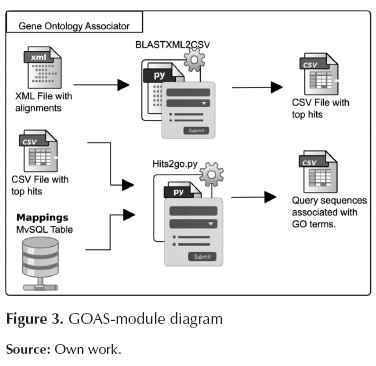
This module involves the following scripts:
BLASTXML2CSV.py: This script selects the best alignment per sequence (top hit) and also writes the new file in CSV format outputting the id from the query sequence associated with the id of the subject sequence with the best alignment score.
Hits2go.py: This script makes an association between sequence identifiers and GO terms using the mappings table provided by the Georgetown University.
GO analyzer
This module categorizes the GO terms according to user's interests. The module also counts how many times particular input sequences appear into the per-user categories and produces a complete report of the results. Additionally, the module is capable of finding sequences that belong to more than one GO category. Figure 4 shows the internal components of this module. The scripts involved in this module are as follows:
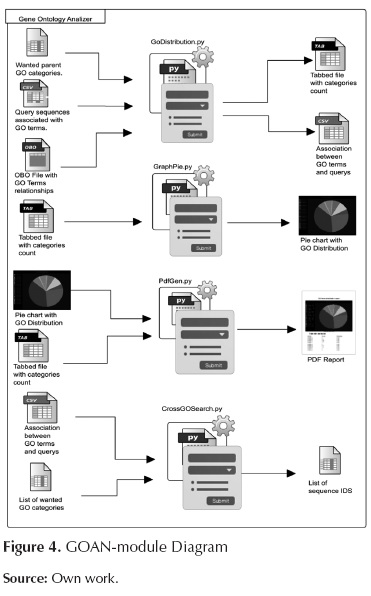
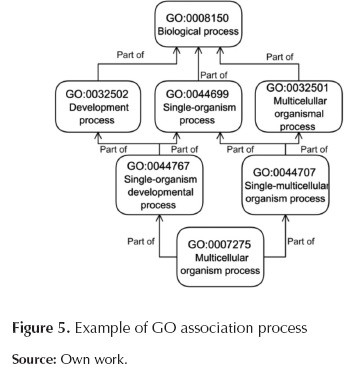
GoDistribution.py: This script associates the desired GO categories (desired by users) to the more abstract GO terms; it also counts how many times input sequences appear per desired GO category. This process is done using the relationship of each GO term with the corresponding parents terms, these relationships has been downloaded previously using AmiGO browser which has been proposed and developed by Carbon et al. (2009). In figure 5 we presented an example explaining how the more specific GO term is associated with a more abstract term. In this case "Multicelullar organism process" will be associated with "Development process", "single-organism process" and "Multicelullar organismal process" categories.
GraphPie.py: This script produces a circular graph that illustrates the distribution of the categories given by the previous analysis.
CrossGOSearch.py: This script is useful to filter all the sequences that appear in various GO categories at the same time, giving to the user the possibility to study a gene that can be involved in various functions at the same time.
PdfGen.py: This script produces a human-readable PDF report that contains the analysis results, this report includes a table with the counts of the GO categories and with the pie chart of the distribution making this software a user-friendly tool.
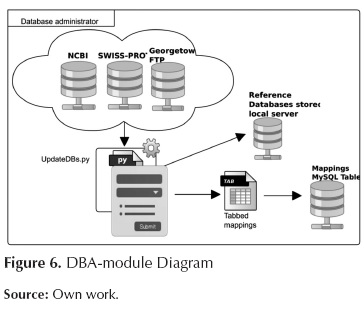
Database administrator
This module carries out updating tasks over the databases of both sequences and mapping so that the databases are available in the local server. This module is presented in figure 6.
The elements involved in this module are as follows:
UpdateDBs.py: Processes: a first process connects to servers NCBI and Swisprot in order to download the databases; another process generates the indices of downloaded files for BLAST, a third process connects to the FTP at Georgetown University in order to download a file that maps the various types of identifiers onto GO terms. This script also executes MappingstoDB.py script which stores the corresponding mapping file in a MySQL table so as to provide quick access.
EVALUATION
Dataset
We have selected a transcriptome FASTA file from Hydractinia Symbiolongicarpus; from the transcriptome we have selected 500, 1000, 2000, 4000 sequences randomly to do the analysis against RefSeq Non-Redundant (Pruitt et al., 2007) and Uniprot (Bairoch et al., (2005) protein databases, both update at May 2014; the expected value selected for the BLAST algorithm is 1e-3.
This transcriptome was selected because it is a representative and typical example of the data normally required to annotate by the researchers from Evolutionary Immunology and Immunogenetics Group from Genetics Institute of Universidad Nacional de Colombia, which are researching for immune response of coral organisms and disappearance of reefs (MAFA was developed in the framework of this research).
Configuration
Processing Cores: 8 out of 24 from a Xeon E7450 @ 2,40GHz
Available RAM: 256GB
Results
Figure 7, figure 8 and table 1 indicate that the module requiring longer processing times is Local Blast Server. Additionally, it can be observed that the relation between processing time and the number of sequences is almost linear, reaching database-dependent rates of 2,7 seconds per processed sequences (for Uniprot) and 50 seconds per processed sequence (for Non-redundant).
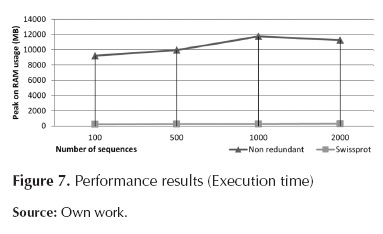
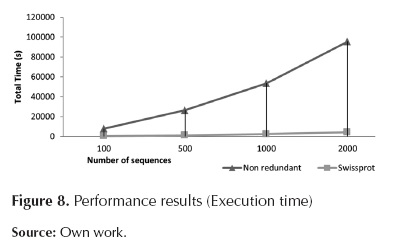
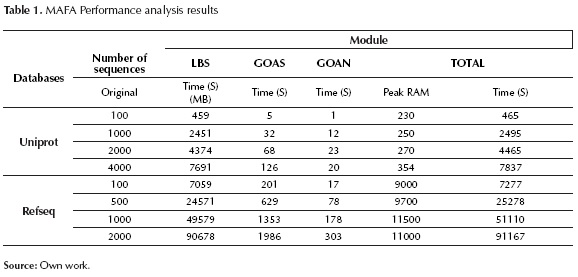
Regarding RAM usage, there is direct dependency on the database in use; on the other hand, there is no dependency on the number of sequences to be processed. For Uniprot, RAM usage is approximately 300MB, for Non-redundant, RAM usage is 11GB.
Conclusions
MAFA is a tool that allows functional annotation and further annotation classification provided there are some given term-specific categories of Gene Ontology. MAFA's main functions include the following: the generation of structured-data outputs that advertise the amount of sequences associated to each GO term, and the establishment of relations between the target term identifiers of Gene Ontology and the identifiers of the given sequences. Additionally, MAFA generates easy-to-interpret graphs for users as well as complete PDF reports containing the results from the corresponding analysis. It is also possible to conduct search processes in order to find sequences that are simultaneously associated to various categories or GO terms.
FUNDING
Center for Scientific Research and Development - Universidad Distrital Francisco José de Caldas.
ACKNOWLEDGEMENTS
Work done in collaboration with High Performance Computational Center (CECAD) - Universidad Distrital Francisco José de Caldas, Bogotá D.C., Colombia (http://cecad.udistrital.edu.co) and Evolutionary Immunology and Immunogenetics Group (http://www.genetica.unal.edu.co/gie/) - Genetics Institute - Universidad Nacional (IGUN), Colombia, (http://www.genetica.unal.edu.co).
References
Altschul. S. F. et al.(1990). Basic local alignment search tool. Journal of molecular biology, 215(3), 403-410. [ Links ]
Ashburner, M. et al. (2000). Gene Ontology: Tool for the unification of biology. Nature genetics, 25(1), 25-29. [ Links ]
Bairoch, A. et al. (2005). The universal protein resource (UniProt). Nucleic acids research, 33(1), D154-D159. [ Links ]
Camacho, C. et al. (2009). BLAST+: Architecture and applications. BMC bioinformatics, 10(1), 421. [ Links ]
Carbon, S. et al. (2009). AmiGO: Online access to ontology and annotation data. Bioinformatics,25(2), 288-289. [ Links ]
Cock, P. J. et al. (2009). Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics,25(11), 1422-1423. [ Links ]
Metzker, M. L. (2010). Sequencing technologies-the next generation. Nature Reviews Genetics,11(1), 31-46. [ Links ]
Pruitt, K. D., Tatusova, T., & Maglott, D. R. (2007). NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic acids research,35(1), D61-D65. [ Links ]

