










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkInfectio
Print version ISSN 0123-9392
Infect. vol.16 no.1 Bogotá Jan./Mar. 2012
1Grupo Inmunovirología, Universidad de Antioquia, Medellín, Colombia
2Grupo de Inmunología Celular e Inmunogenética, Instituto de Investigaciones Médicas, Universidad de Antioquia, Medellín, Colombia
Recibido: 27/10/2011; Aceptado: 08/02/2012
Resumen
El monofosfato de adenosina cíclico (AMPc) induce la activación de la proteína cinasa A, la cual regula negativamente la activación, la proliferación celular y la producción de IL-2, en células T. En células infectadas con el virus de inmunodeficiencia humana, el monofosfato de adenosina cíclico suprime la actividad de transcripción del promotor del virus y el paso del ADN viral del citoplasma al núcleo. El incremento del monofosfato de adenosina cíclico mediado por células T reguladoras CD4+, empleando la inyección de esta molécula en células blanco a través de las uniones comunicantes o empleando el eje CD39-CD73 para generar adenosina es utilizado para suprimir otras poblaciones celulares.
En esta revisión se propone que la modulación del monofosfato de adenosina cíclico por las células T reguladoras CD4+ podría tener un papel dual durante la evolución de la infección por el virus de inmunodeficiencia humana. Su papel benéfico se centraría principalmente en el control de la replicación viral y factores de transcripción, o evitando la infección de nuevas células blanco por disminución en la expresión de los receptores virales. Paradójicamente, la segunda posibilidad es que el aumento del monofosfato de adenosina cíclico podría tener un papel perjudicial, debido al efecto negativo sobre la proliferación, activación, respuesta citotóxica y en la producción de citocinas que se observa durante la infección viral.
Palabras clave: AMP cíclico, VIH, células T, replicación viral, uniones comunicantes, adenosina.
Abstract
Cyclic adenosine monophosphate induces the activation of protein kinase A, which negatively regulates activation, proliferation and IL-2 production in T cells. In cells infected with human immunodeficiency virus, cyclic adenosine monophosphate suppresses the transcriptional activity of long terminal repeats and the amount of viral DNA from the cytoplasm to the nucleus. The increase in cyclic adenosine monophosphate mediated by CD4+ regulatory T cells, using either the influx of this molecule in target cells through the GAP junctions or by CD39-CD73 to generate adenosine, is used by CD4+ regulatory T cells to suppress other cell populations. In this review, we suggest that modulation of cyclic adenosine monophosphate by CD4+ regulatory T cells may have a dual role during the evolution of human immunodeficiency virus infection. The beneficial role would be mainly focused on the control of viral replication and transcription factors to replicate the virus, and/or preventing the infection of new target cells, decreasing the expression of the viral co-receptors. Paradoxically to this beneficial role, the second possibility is that increased cyclic adenosine monophosphate could have a detrimental role, due to the negative effect on proliferation, activation, cytotoxic response and cytokine production, which occurs during viral infection.
Key words: Cyclic AMP, HIV, T cells, virus replication, Gap junctions, adenosine.
Introducción
El monofosfato de adenosina cíclico (AMPc) es un segundo mensajero involucrado en la actividad de transcripción de genes que median la progresión del ciclo celular, diversas respuestas celulares y las respuestas inmunitarias innata y adaptativa. Esta molécula puede regular la actividad funcional de células T reguladoras CD4+Foxp3+ (Treg), T convencionales CD4+Foxp3- (Tcon) y de células presentadoras de antígeno (1,2). A diferencia de las células Tcon, las Treg CD4+ tienen un papel crucial en la supresión de la respuesta inmunitaria, controlando la respuesta inflamatoria exacerbada, los procesos autoinmunitarios y las respuestas alérgicas (3).
En algunos estudios se ha resaltado la importancia del AMPc en las alteraciones inmunitarias asociadas a la infección por el virus de la inmunodeficiencia humana (VIH) (4-6), particularmente, por ser uno de los mecanismos de supresión de las Treg (2,4). Igualmente, se ha postulado que mediante la activación de la vía del AMPc se pueden controlar los procesos de infección y replicación del virus (7,8).
La infección con el VIH se caracteriza por el desarrollo progresivo de una inmunodeficiencia que compromete la inmunidad innata y la adaptativa. Estas alteraciones funcionales incluyen disminución en la producción de citocinas, proliferación de células T y disminución de la citotoxicidad de células T CD8+, así como una disminución en la maduración y producción de interleucina 12 (IL-12) por células dendríticas (9,10). Las alteraciones se han asociado con cambios cuantitativos y funcionales de las células T CD4+, con la supresión mediada por las Treg CD4+, y con la expresión de moléculas inhibitorias en diferentes subpoblaciones celulares.
En esta revisión se describen los aspectos relacionados con el uso del AMPc por las células Treg CD4+, como mecanismo de regulación inmunitaria, y el efecto del AMPc en la modulación de la infección-replicación del VIH. Para el desarrollo de esta revisión, se buscó en las base de datos Pubmed, combinando los términos: HIV-1 and cAMP, regulatoryTcells and GAP junctions, CD39, adenosine.
Vía de señalización del AMPc
Los niveles de AMPc intracelular son controlados por dos grupos de enzimas, la adenilciclasa, que se encuentra en su mayoría unida a la cara interna de la membrana celular, que usa el trifosfato de adenosina (ATP) como sustrato para producir AMPc, y por las fosfodiesterasas, ubicadas en diferentes compartimientos subcelulares, que hidrolizan el AMPc hacia su forma inactiva, conocida como adenosina 5`-monofosfato (11). En células de mamíferos, se han reportado hasta el momento 10 diferentes isoformas de la familia de enzimas de la adenilciclasa (AC1-AC10), y 11, para la fosfodiesterasa (PDE1-PDE11) (12-15).
El incremento del AMPc induce la activación de la proteína cinasa A, la cual regula la activación de las células T y la transcripción de los genes involucrados en la progresión del ciclo celular, las vías glucolíticas y las lipolíticas (16). La unión del AMPc a la subunidad reguladora de la proteína cinasa A induce su activación al liberar la subunidad catalítica; en la cara interna de la membrana celular, la fosforilación de tirosina cinasa C-src (Csk) por la proteína cinasa A incrementa su actividad; posteriormente, la Csk fosforila e inactiva a la tirosina cinasa específica de linfocitos (Lck), proteína importante en la activación proximal del receptor de células T (17). Diferentes vías de señalización pueden ser reguladas por la actividad de la proteína cinasa A; la proteína unidora de los elementos de respuesta al AMPc (CREB) es fosforilada por esta cinasa en la serina 133, lo cual bloquea la formación del complejo con el coactivador de unión a CSK (CBP) y la unión a los elementos de respuesta al AMPc (CRE) (18), los cuales pueden ser encontrados en genes que codifican por el receptor de células T o en otros genes involucrados en la activación de la células T (19). Además, la proteína cinasa A regula la actividad del factor nuclear de células T activadas. Cuando esta proteína es fosforilada por la proteína cinasa A, crea sitios de unión para otro proteína llamada 14-3-3 (grupo de proteínas diméricas altamente conservadas); la formación de este nuevo complejo disminuye la actividad de transcripción del factor nuclear de células T activadas (20).
En estado reposo, el NF-κB se encuentra en el citoplasma de la célula acoplado a su inhibidor IκB, que previene su translocación al núcleo. Durante la activación celular, el IκB es fosforilado por una cinasa de IKB, lo cual induce la separación del complejo NF-κB/IκB (21). Por el contrario, cuando la proteína cinasa A es fosforilada, su subunidad catalítica (PKA-C) se une a este complejo NF-κB/IκB, estabilizándolo y manteniendo el complejo inactivo (21).
Otros blancos de fosforilación de la proteína cinasa A son las proteínas Raf-1, Ras, Mek y HePTP de la vía de las MAPK cinasas, y de PLC-α1 o PLC-β, en la vía del fosfatidil-inositol (17,21).
En resumen, esto sugiere que la señalización inducida por la proteína cinasa A bloquea las interacciones entre proteínas y la actividad enzimática de diferentes cinasas. Entre los sustratos de la proteína cinasa A involucrados en la activación inmunitaria, se incluyen moléculas tempranas de la activación celular, tardías como factores de transcripción y, además, miembros de las MAPK cinasas y fosfolipasas.
Aunque el principal blanco del AMPc es la proteína cinasa A, también se ha demostrado que activa directamente a la proteína intercambiadora activada por AMPc (EPAC-1 y 2). Esta vía es independiente de la proteína cinasa A y regula la activación de una GTPasa llamada Rap-1 (22). Esta se ha asociado al mantenimiento de la anergia de las células T y a la regulación negativa de genes de citocinas, moléculas coestimuladoras y receptores de quimiocinas (22,23).
Diferencias metabólicas que definen los niveles de AMPc en células Treg CD4+ y células Tcon
Las Treg son una subpoblación de células T CD4+ caracterizadas por ser potentes inhibidores de la activación y expansión de otras subpoblaciones celulares, tanto in vitro como in vivo. Las células Treg CD4+, además de requerir del estímulo del receptor de células T, son dependientes de la vía de señalización inducida por la IL-2 para su desarrollo, expansión y función en la periferia. Clásicamente, estas células se identifican por la ausencia o baja expresión del receptor de la IL-7 (CD127), por la expresión o alta densidad de la cadena alfa del receptor de la IL-2 (CD25) y el factor de transcripción Foxp3 (CD4+CD127-/ LowCD25+/HiFoxp3+)(24). Las células Treg CD4+ se clasifican en Treg naturales, si se originan directamente en el timo, o Treg inducidas, si se originan en la periferia a partir de conversión de una célula Tcon (25). Además, estas células Treg pueden expresar otras moléculas, como el antígeno 4 del linfocito T citotóxico (CTLA-4) y el receptor del factor de necrosis tumoral inducido por glucocorticoides (GITR), los cuales están asociados a su actividad supresora (25,26). Por su lado, una célula Tcon CD4+ o CD8+ se caracteriza por la ausencia de expresión de Foxp3 y por su capacidad para llevar a cabo funciones efectoras.
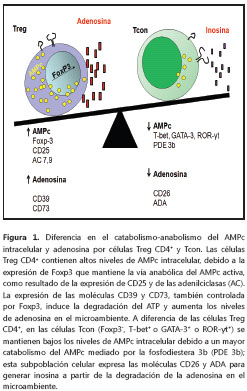
A diferencia de las Tcon, las células Treg CD4+contienen grandes cantidades de AMPc (2) (figura 2), lo cual se puede explicar por la alta expresión, 50 veces más, de la adenilciclasa 9 (AC9) (27). Además, se ha observado que la alta expresión del CD25 en las células Treg CD4+, lo cual les confiere una ventaja competitiva por la IL-2 con respecto a las Tcon (28), favorece la activación de la adenilciclasa 7 y la acumulación de AMPc (29); esta expresión diferencial del CD25 y las adenilciclasas en las células Treg CD4+ es controlada por el Foxp3. En contraste con este efecto anabólico, las Treg CD4+ tienen una baja tasa de degradación del AMPc, debido a que se ha observado que en esta subpoblación celular la expresión de la enzima fosfodiesterasa 3b está disminuida (figura 1) (30,31). Hallazgos recientes demuestran que la concentración de AMPc es controlada por el miR-142-3p, un microARN que regula la actividad de transcripción del Foxp3 y, por ende, la expresión de la adenilciclasa; en las células Tcon, el miR-142-3p inhibe la producción de adenilciclasa 9, mientras que en las Treg CD4+ este efecto no se observa, debido a que el factor de transcripción Foxp3 regula negativamente la expresión de miR-142- 3p y mantiene activa la vía AC9/AMPc (27). Estos datos sugieren que la gran actividad anabólica mediada por la adenilciclasa con la menor actividad catabólica mediada por fosfodiesterasas, explicarían los altos niveles de AMPc en las células Treg CD4+ (figura 1).

El AMPc en procesos de regulación inmunitaria
Alteraciones en la respuesta inmunitaria mediadas por el incremento de AMPc intracelular. En células del sistema inmunitario, la vía AMPc/PKA modula la proliferación y la transcripción de genes de citocinas a través de varias vías de señalización (1,17,32). El tratamiento de las células Treg CD4+con rolipram, un inhibidor de la fosfodiesterasa E4 (PDE4), previene la poca degradación del AMPc intracelular, incrementando aún más los niveles de AMPc y potenciando su capacidad supresora in vivo sobre células Th2; de esta manera, se logra un mayor control de la inflamación tisular y de las enfermedades respiratorias alérgicas (33). Por el contrario, en las células Treg CD4+ de pacientes con cáncer de colon, el tratamiento con antagonistas (análogos estructurales del AMPc, inhibidor competitivo por el sitio de unión a la proteína cinasa A) disminuye la supresión inmunitaria y aumenta la respuesta inmunitaria antitumoral (34), lo cual sugiere que la inhibición de la vía AMPc/PKA podría ser un blanco terapéutico en enfermedades en las que la excesiva regulación inmunitaria mediada por las células Treg CD4+ juega un papel patogénico. Cuando las células Tcon son tratadas independientemente con la toxina del cólera, ésta actúa sobre la subunidad alfa de la proteína G heterotrimérica (responsable de traducción de señales intracelulares), catalizando la unión de ADP-ribosa citoplásmica a dicha subunidad, reacción llamada “ribosilación” de ADP. Esta unión produce la activación permanente de la adenilciclasa, la que a su vez aumenta los niveles de AMPc, lo cual está asociado con la disminución en la proliferación y en la producción de IL-2 en respuesta a la estimulación con anti-CD3. Además, este estímulo promueve la adquisición de funciones reguladoras en células Tcon, como la inhibición de la proliferación de otras Tcon (en cocultivos de células Tcon tratadas con la toxina del cólera y Tcon sin tratar) (35). El mecanismo propuesto para este efecto es el aumento en la expresión de la molécula inhibitoria CTLA-4, mediado por el AMPc (36). Esto indica que se necesitan bajas o altas concentraciones de AMPc en células Tcon para definir su función efectora o supresora, espectivamente.
Además, el AMPc puede afectar la actividad funcional de los monocitos y de las células dendríticas; éste inhibe la maduración de estas subpoblaciones celulares, así como la capacidad de expresar CD40 (molécula coestimuladora), la internalización de antígenos (37) y la capacidad de producir el factor de necrosis tumoral alfa (TNF-α) (38), IL-12 p35 y p40, al bloquear la unión del factor de transcripción IRF-8 al promotor de la IL-12 (39). Otra citocina regulada por el AMPc es la IL-10, la cual controla en células dendríticas la expresión de moléculas coestimuladoras del complejo mayor de histocompatibilidad de clase II (el cual contiene elementos CRE) y la producción de IL-12 (38-42). En monocitos, se ha reportado que el AMPc induce la producción de IL-10, por medio de la activación de los factores de transcripción CREB-1 y ATF-1(43). Esto sugiere que el AMPc afecta de forma directa la habilidad funcional de las células T, pero, indirectamente, también compromete la habilidad de las APC para estimular células Tcon y modificar el microambiente de citocinas para inhibir la diferenciación de células Th1.
La activación de la enzima adenilciclasa inducida por las prostaglandinas se produce principalmente a través de receptores acoplados a proteínas Gαs, ancladas a la membrana celular. Mediante este mecanismo, la prostaglandina I2 (PGI2) en células dendríticas derivadas de monocitos incrementa los niveles de AMPc y regula negativamente la actividad del NF-kB, disminuyendo la producción de citocinas proinflamatorias, como IL- 12, TNF-α, IL-1α e IL-6, e incrementando la producción de IL-10 (44). La prostaglandina E2 (PGE2) inhibe la producción de interferón alfa (IFN-α) en células dendríticas plasmacitoides (CDp) y la producción de IL-12 en células dendríticas mieloides (CDm) en respuesta a estímulos virales y a lipopolisacáridos, respectivamente (45). La prostaglandina E2 suprime la proliferación celular e inhibe la producción de IL-2 e interferón gamma (IFN-γ) en células T de ratón o humanas (46).
La gran mayoría de las alteraciones en la respuesta inmunitaria son mediadas por la vía AMPc/PKA. Sin embargo, también se han observado alteraciones mediante un mecanismo independiente de la proteína cinasa A, el cual involucra la vía EPAC/RAP-1 para la supresión de la respuesta inmunitaria. Esta última vía regula negativamente la expresión de algunos genes asociados con el ciclo celular, limitando la proliferación de células T, e igualmente, disminuyendo la producción de citocinas como IL-2 e IL-5 (22, 47, 48). De hecho, la expresión de la forma activa de RAP-1 se ha observado en células T anérgicas y se considera como un regulador negativo de la transcripción génica inducida por el receptor de células T y la IL-2 (49). En células dendríticas, la señalización de EPAC al parecer no tiene ningún efecto en la maduración y función (50).
Alteraciones en la respuesta inmunitaria inducida por células Treg CD4 + mediante la modificación del AMPc intracelular
Las células Treg CD4+ median la supresión de otras células del sistema inmunitario por el aumento de AMPc mediante dos mecanismos diferentes:
1. La CD39 y la CD73 son moléculas pertenecientes a una familia de enzimas llamadas ectonucleotidasas; CD39 y CD73 se expresan constitutivamente en el 70 a 80 % de las células Treg CD4+ y, una en una menor proporción (8 a 15 %), en células Tcon (51). Las células Treg CD4+, CD39+ y CD73+ aumentan la adenosina en el microambiente, la cual es reconocida por el receptor de adenosina 2 en la membrana celular, incrementando el AMPc intracelular en las Tcon (figura 2) (52). El ATP extracelular es un factor proinflamatorio generado en condiciones de estrés y durante el daño celular; de hecho, se considera un indicador de destrucción celular. Para controlar los niveles excesivos de ATP, la CD39 en la superficie de las células Treg CD4+ hidroliza el ATP a 5-AMP, y actúa en conjunto con otra ecto-enzima, la CD73, también expresado en las Treg CD4+, generando adenosina a partir del 5-AMP (figura 2C) (52). La adenosina pericelular se une al receptor de adenosina 2 sobre las células Tcon activadas y esta interacción (adenosina-receptor de adenosina 2) induce la activación de la proteína G que se encuentra acoplada a la porción intracelular del receptor, lo que finalmente promueve la activación de la adenilciclasa y el incremento de AMPc intracelular (figura 2C) (52). Como mecanismo antagónico de este proceso, se encuentra la acción de la enzima desaminasa de adenosina (ADA), que degrada la adenosina a inosina. Esta enzima se asocia a la porción extracelular de la molécula CD26, que tiene función de dipeptidil peptidasa IV, y ambas moléculas participan en el catabolismo de la adenosina pericelular (53). La molécula CD26 promueve la activación de células T o bien la interacción entre éstas y las células presentadoras (53). La generación de inosina por parte de ADA-CD26 es reconocida posteriormente por el receptor de adenosina A2, el cual no induce aumento del AMPc intracelular y activación de la proteína cinasa A, ni tiene efectos posteriores a la transcripción que involucren la vía de las MAPK cinasas o del NF-κB (54,55). Esta inosina disminuye la producción de citocinas proinflamatorias por parte de monocitos y macrófagos (55-57), e incrementa la degranulación de mastocitos (58).
La CD39 fue inicialmente descrita como una molécula expresada sólo durante la activación; sin embargo, actualmente se considera como una molécula reguladora, cuya expresión es regulada por el Foxp3 (59), así como por la señalización mediada por el AMPc/ PKA que induce la fosforilación de CREB1 y ATF-2 (60), lo cual sugiere que entre el AMPc y la CD39 se podría generar una retroalimentación recíproca, para su expresión constante.
El eje CD39-CD73 es empleado por las células Treg CD4+como mecanismo de supresión para controlar la respuesta inflamatoria y la respuesta efectora de células T (59,61). Estas células bloquean la proliferación de células T, la producción de IFN-γ y la maduración de células dendríticas por el incremento de la adenosina en el espacio extracelular (59,62). En las células Tcon, la adenosina o compuestos agonistas del receptor de adenosina 2 disminuyen la producción de las citocinas proinflamatorias IFN-γ y TNF-α de células T, y la citotoxicidad de células T CD8+ (63-65).
Es probable que el desarrollo de una respuesta reguladora o inflamatoria sea determinada por la presencia de la adenosina, ya que los niveles de esta molécula son controlados por el equilibrio generado por las células Treg CD4+ y las Tcon, en el cual las primeras se encargan de aumentar los niveles de adenosina con la expresión del CD39HiCD73HiADALowCD- 26Low, mientras que las segundas se encargan de su degradación con la expresión del CD- 39LowCD73LowADAHiCD26Hi (figura 2) (51).
2. Las células Treg CD4+ introducen el AMPc en la células blanco mediante comunicaciones intercelulares llamadas uniones comunicantes (gap junctions) (figura 3C) (2). Las uniones comunicantes son canales intercelulares que permiten la comunicación entre células adyacentes. Están formadas por dos hemicanales opuestos de cada célula, llamados conexones, y constituidas por seis proteínas llamadas conexinas. Cada conexina tiene un tamaño aproximado que varía entre 26 y 60 kDa. Están formadas por cuatro dominios transmembrana, con una región N y una C terminal citoplásmica; estas cuatro regiones están conectadas entres sí por dos asas extracelulares y una intracelular (66). En células de mamífero, las uniones comunicantes son utilizadas para el paso bidireccional de iones, metabolitos y otras moléculas menores de 1 kDa (67,68). Las células Treg CD4+ y las Tcon, en condiciones basales, exhiben una baja densidad de expresión de las conexinas Cx31.1, Cx32, Cx43, Cx45 y Cx46, las cuales se incrementan con la activación celular (2).
Como se comentó anteriormente, las T contienen bajos niveles de AMPc intracelular, el cual se incrementa cuando son cocultivadas con células Treg CD4+, disminuyendo así la expresión de IL-2 en las Tcon (2). En ensayos con calceína, un colorante que es transferido a través de las uniones comunicantes, y en ensayos de bloqueo con el péptido sintético GAP27, el cual inhibe la Cx43 y afecta la formación y la estabilidad de las uniones comunicantes (66), se demostró que las células Treg CD4+ suprimen a las Tcon mediante la transferencia de moléculas a través de dichas uniones (2); experimentos posteriores permitieron demostrar la participación del AMPc como la molécula involucrada en este mecanismo de supresión (68,69).
El incremento de AMPc mediado por células Treg CD4+ mediante las uniones comunicantes no sólo ocurre hacia las Tcon, como inicialmente fue reportado por Bopp et al. (2007) (2), sino que también se ha descrito en otras subpoblaciones celulares que son blanco de la supresión, tanto in vivo como in vitro. Recientemente, también se reportó que las células Treg CD4+ afectan la habilidad de las células dendríticas para presentar antígenos, debido a que el AMPc transferido desde las primeras disminuye la expresión de las moléculas coestimuladoras CD80 y CD86, y la secreción de IL-6 y de IL-12, mientras que aumenta la expresión de moléculas inhibidoras como B7-H4 y la producción de IL-10 (70,71).
Papel dual del AMPc en la respuesta inmunitaria y la replicación viral durante la infección con VIH
La mayoría de datos sugieren que el AMPc tiene un efecto negativo en el desarrollo de la respuesta inmunitaria, al inhibir la transcripción de genes asociados con el desarrollo de esta respuesta. Sin embargo, durante la infección por el VIH se ha observado un papel dual de esta molécula, ya que no sólo suprime la respuesta inmunitaria antiviral, sino que también está asociado a un efecto protector, al inhibir la replicación viral (figura 2).
Alteraciones en la respuesta inmunitaria asociadas al incremento de AMPc intracelular durante la infección con el VIH.
El tratamiento de células T CD3+ de pacientes positivos para VIH con agonistas del AMPc, induce la regulación positiva de 8 genes y la negativa de 144 genes, lo cual modifica la expresión de varias integrinas, quimiocinas y receptores de quimiocinas (32). En diversos ensayos ex vivo se ha demostrado que las células T de pacientes positivos para VIH exhiben concentraciones de AMPc dos veces mayores que las de individuos negativos para VIH, así como una actividad mayor de la proteína cinasa A, lo que se correlaciona inversamente con la capacidad de proliferación en respuesta a antígenos de Candida albicans y en respuesta a la activación inducida con anti-CD3 (6). Durante la infección por el VIH, se demostró que las células provenientes de pacientes positivos para VIH sin tratamiento antirretroviral, exhiben un incremento en la actividad ATPasa, fenómeno que se asoció con un mayor porcentaje de células T CD39+ totales, en comparación con controles sanos (72). Recientemente, se reportó una correlación positiva entre la expresión de la CD39 en células Treg CD4+ con la carga viral y la progresión de la enfermedad (4) .En otros modelos en que se emplearon primates infectados con el virus de inmunodeficiencia simiana, se demostró que las células T CD8+Foxp3+CD25+ tienen una mayor expresión de CTLA-4 y CD39 en los sitios de mayor replicación viral, como la mucosa gastrointestinal y los órganos linfoides, este fenotipo celular se asoció con una disminución en el control viral y con la supresión de células T específicas de antígenos virales (73).
Mecanismos de incremento de AMPc intracelular inducido por el virus o las proteínas virales. El incremento del AMPc induce anergia de células T, mediante la unión de la glucoproteína 120 (gp120) con la molécula CXCR4, lo cual induce el incremento y la activación de la vía de señalización AMPc/PKA (74,75). Este mecanismo involucra la fosforilación de CREB por la proteína cinasa A, lo cual resulta en la reducción de la proliferación de células T en respuesta a estímulos policlonales (65,66). La inhibición en la generación de AMPc intracelular por compuestos químicos, como el análogo de adenosina (2',5'-dideoxiadenosina, ddADA) que inhibe la adenilciclasa, restaura la capacidad citotóxica y de proliferación de las células T (5,74). La interacción de la glucoproteína 120 con la molécula CD4 en células Treg CD4+, también incrementa los niveles de AMPc, lo que se correlaciona con un aumento en la expresión de la molécula CTLA-4 (76). Infortunadamente, aún se desconoce si las alteraciones en los niveles de AMPc en pacientes positivos para VIH están asociadas a la acumulación de células Treg CD4+ durante la infección crónica, lo cual podría afectar el desarrollo de una adecuada respuesta inmunitaria antiviral (77).
En pacientes positivos para VIH, la actividad de la enzima desaminasa de adenosina (encargada de mantener niveles bajos de adenosina) puede incrementar la proliferación y la producción de IFN-γ en células T; de hecho, hay una correlación positiva entre la desaminasa de adenosina y el conteo de células T CD4+, y una correlación negativa con la carga viral (78). Sin embargo, es importante aclarar que durante la infección por el VIH el complejo CD26-ADA desaminasa de adenosina es alterado por proteínas virales, el cual es mediado por la interacción del CD4 o CXCR-4 con la glupoproteína 120 (79, 80) y por la proteína viral Tat, producida intracelularmente en células infectadas que inhiben la unión de la desaminasa de adenosina con el CD26 (81).
Efecto del incremento de AMPc intracelular en la replicación del VIH y su relación con las células Treg CD4+
En cultivos primarios y en líneas de células T humanas infectadas con el VIH, el AMPc es una molécula involucrada en la supresión de la actividad de transcripción del promotor viral (LTR, repeticiones terminales largas) al inhibir NF-kB, reduciendo de esta manera la replicación viral (8). Además, el AMPc disminuye la importación del ADN viral del citoplasma al núcleo; en células T vírgenes la replicación y translocación de ADN viral al núcleo son significativamente menores en comparación con las de las células T de memoria, lo cual es debido a la baja expresión de fosfodiesterasa 4, que promueve una mayor acumulación de AMPc intracelular en las células vírgenes (7). Esto sugiere que la vía AMPc/PKA puede modular los estados de previos y posteriores a la integración viral (7). Infortunadamente, el efecto de la activación de la vía AMPc/EPAC sobre la replicación del VIH no se conoce hasta el momento.
Los compuestos sintéticos que incrementan el AMPc mediante la activación de la adenilciclasa, como la forskolina (Fsk), o el bloqueo con el inhibidor selectivo de la fosfodiesterasa 4 con rolipram, reducen los niveles de la proteína viral p24 y la transcripción del provirus en cultivos de células T activadas(7, 82). La activación de la enzima adenilciclasa por la prostaglandina E2 (83), incrementa el AMPc intracelular y controla la replicación del VIH al disminuir la actividad de las repeticiones terminales largas, lo cual se ha observado tanto en macrófagos primarios como en líneas celulares crónicamente infectadas (84). El AMPc también podría prevenir la infección por el virus en monocitos y macrófagos, al menos de cepas R5, ya que el estímulo del receptor de la prostaglandina E2 disminuye la expresión del CCR5, el cual es indispensable para la entrada eficiente del virus a su célula blanco (85).
Durante la infección por el VIH se ha reportado un efecto benéfico de la vía adenosina-AMPc; el tratamiento con ATP induce la degradación lisosómica del virus en células dendríticas inmaduras (CDi) y también bloquea el tráfico de viriones desde estas últimas a las células T CD4+ (86). También, a través de la estimulación del receptor de adenosina 2 con anticuerpos monoclonales, se puede controlar la entrada del virus a la célula blanco por la disminución en la expresión de los correceptores CXCR-4 y CCR-5 en células T CD4+ (87). En células T CD4+ vírgenes, se ha observado que el tratamiento con el factor derivado de células del estroma, activa la vía AMPc-PKA-CREB y altera la capacidad de proliferación de estas células, pero al mismo tiempo, disminuye la expresión del correceptor viral CXCR4(75).
Debido a que el AMPc reduce la replicación viral y a que las células Treg CD4+ pueden inyectar esta molécula a través de las uniones comunicantes en la célula blanco, o modificar los niveles de adenosina en el microambiente, recientemente exploramos el papel de esta subpoblación celular durante la infección por el VIH. En ensayos in vitro de un modelo de infección aguda, las células Treg CD4+ redujeron en un 50 % el porcentaje de las células Tcon infectadas con el VIH y, también, los niveles de p24 en los sobrenadante de cocultivos de células Treg CD4+ y Tcon (69). Los ensayos con calceína para evaluar la interacción entre células Treg y Tcon, junto con la de separación de las dos subpoblaciones con membranas permeables transwell y el bloqueo de la formación de las uniones comunicantes con el inhibidor GAP27, sugirieron que el mecanismo de supresión depende del contacto entre las células (69). Posteriormente, la inhibición de las enzimas adenilciclasa y ectonucleotidasa CD39, con ddADA y anti-CD39, respectivamente, corroboró el papel crucial que tienen los niveles altos de AMPc y la generación de adenosina en la supresión de la replicación del VIH ejercida por las células Treg (69).
Para dilucidar los mecanismos implicados en la supresión mediada por AMPc en la replicación del VIH y la contribución de la proteína cinasa A como efector corriente abajo de la actividad de AMPc, se llevaron a cabo ensayos con un inhibidor de la proteína cinasa A con el H89 diclorhidrato, el cual compite con el ATP por la unión al bolsillo de la subunidad catalítica de la cinasa y con el éster acetoximetil- de N6-benzoil-AMPc (6-BnzcAMP- AM), análogo del AMPc y activador específico de la proteína cinasa A. Los resultados sugirieron que las células Treg median la activación de la proteína cinasa A mediante el AMPc para controlar la replicación viral (69).
Finalmente, toda esta cascada de señalización disminuye la proliferación y el ciclo celular (Ki67, ciclina B) en las células Tcon, lo que se refleja en la disminución en el número de células infectadas. En resumen, todos estos hallazgos indican que las células Treg podrían tener un papel benéfico in vivo durante la fase aguda de esta infección, particularmente en los órganos de mayor activación inmunitaria. Sin embargo, es necesario confirmar esto en otros modelos en pacientes positivos para VIH o primates infectados por el VIS. Igualmente, se debe evaluar el papel positivo o negativo del AMPc y su asociación con las Treg CD4+ durante las distintas fases de infección por el VIH.
Conclusiones
El incremento del AMPc modula la activación de células T y la transcripción de genes involucrados en la proliferación celular, citocinas, quimiocinas y diferentes receptores de superficie a través de varias vías de señalización intracelular. Esta molécula es utilizada por las células Treg CD4+ dentro de su arsenal de mecanismos para suprimir la respuesta inmunitaria. El papel del AMPc durante la infección por el VIH ha sido motivo de varias investigaciones, con diferentes resultados contradictorios. Sin embargo, de acuerdo con los hallazgos presentados hasta el momento, nosotros proponemos que el AMPc podría tener un papel dual durante la evolución de la infección por el VIH. Su papel benéfico se centraría principalmente en el control de la replicación viral, bloqueando la actividad de las repeticiones terminales largas y factores de transcripción, o evitando la infección de nuevas células blanco, por disminución en la expresión de los correceptores virales como CCR5 y CXCR4.
Uno de los mecanismos que podría explicar este fenómeno es el incremento de AMPc mediado por células Treg CD4+, ya sea empleando la inyección de esta molécula en células blanco o a través del eje CD39-CD73 para generar adenosina. Sin embargo, los datos son escasos y no permiten establecer claramente el papel de estos mecanismos de supresión viral. Paradójicamente con este papel benéfico, la segunda posibilidad es que el aumento de AMPc podría tener un papel perjudicial. De hecho, se considera que el efecto negativo sobre la proliferación, activación, respuesta citotóxica y en la producción de citocinas, que se observa durante la infección viral, se asocia con altos niveles de AMPc. La supresión inmunitaria mediada por el incremento de AMPc (el cual puede ser inducido por proteínas virales) o por la expansión de las células Treg CD4+ (las cuales emplean este mecanismo de AMPc de forma inespecífica), es una estrategia utilizada por el virus para favorecer el establecimiento de la infección y evitar su eliminación.
En conclusión, el papel dual que se observa con el AMPc, también estaría subordinado al tipo de célula en la cual está regulando la actividad de transcripción. El AMPc podría tener un efecto protector, principalmente en células infectadas, pero, en contraste, tendría un papel perjudicial en células del sistema inmunitario responsables de la eliminación de las células infectadas. Además, el papel de las células Treg CD4+ podría cambiar según la fase de la infección. Durante la infección aguda por el VIH, esta subpoblación celular podría proteger a los individuos infectados, controlando la excesiva inflamación y la activación inmunitaria, Sin embargo, en la fase crónica, la expansión de estas células podría tener un efecto deletéreo al suprimir la respuesta inmunitaria adaptativa. Finalmente, es importante diseñar más estudios que permitan evaluar el papel de la vía AMPc/AC/PKA, de la adenosina, del receptor de adenosina 2 y de las uniones comunicantes, así como de sus antagonistas en células del sistema inmunitario durante la infección por el VIH, con el objetivo de establecer nuevos blancos terapéuticos del tratamiento antirretroviral o identificar posibles moléculas dianas con potencial inmunorregulador que permitan restablecer la disfunción inmunitaria.
Agradecimientos
A Colciencias por la financiación del proyecto “Role of regulatory T cells in gut-associated lymphoid tissue during type 1 human immunodeficiency virus (HIV-1) infection” (111540820490-1); César Mauricio Rueda es financiado por Colciencias en el programa de doctorados nacionales.
Conflicto de intereses
Los autores no tienen conflictos de intereses.
Correspondencia: María Teresa Rugeles, Calle 62 N° 52-59, oficina 532, Sede de Investigación Universitaria, Medellín, Colombia.
Teléfono: (574) 219-6482; fax: (574) 219-6481. Dirección electrónica: mtrugel@udea.edu.co
Referencias
1. Gerlo S, Verdood P, Kooijman R. Modulation of cytokine production by cyclic adenosine monophosphate analogs in human leukocytes. J Interferon Cytokine Res. 2010;30:883-91. [ Links ]
2. Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med. 2007;204:1303-10. [ Links ]
3. Bacchetta R, Gambineri E, Roncarolo MG. Role of regulatory T cells and FOXP3 in human diseases. J Allergy ClinImmunol. 2007;120:227-35. [ Links ]
4. Schulze J, Thomssen A, Hartjen P, Toth I, Lehmann C, Meyer-Olson D, et al. Comprehensive analysis of frequency and phenotype of T regulatory cells in HIV infection: CD39 expression of FoxP3+ T regulatory cells correlates with progressive disease. J Virol. 2010;85:1287-97. [ Links ]
5. Hofmann B, Nishanian P, Nguyen T, Liu M, Fahey JL. Restoration of T-cell function in HIV infection by reduction of intracellular cAMP levels with adenosine analogues. AIDS. 1993;7:659-64. [ Links ]
6. Aandahl EM, Aukrust P, Skalhegg BS, Muller F, Froland SS, Hansson V, et al. Protein kinase A type I antagonist restores immune responses of T cells from HIV-infected patients. Faseb J. 1998;12:855-62. [ Links ]
7. Sun Y, Li L, Lau F, Beavo JA, Clark EA. Infection of CD4+ memory T cells by HIV-1 requires expression of phosphodiesterase 4. J Immunol. 2000;165:1755-61. [ Links ]
8. Banas B, Eberle J, Schlondorff D, Luckow B. Modulation of HIV- 1 enhancer activity and virus production by cAMP. FEBS Lett. 2001;509:207-12. [ Links ]
9. Boasso A, Shearer GM, Chougnet C. Immune dysregulation in human immunodeficiency virus infection: know it, fix it, prevent it? J InternMed. 2009;265:78-96. [ Links ]
10. Martinson JA, Román-González A, Tenorio AR, Montoya CJ, Gichinga CN, Rugeles MT, et al. Dendritic cells from HIV-1 infected individuals are less responsive to toll-like receptor (TLR) ligands. Cell Immunol. 2007;250:75-84. [ Links ]
11. Omori K, Kotera J. Overview of PDEs and their regulation. Circ Res. 2007;100:309-27. [ Links ]
12. Beavo JA. Cyclic nucleotide phosphodiesterases: Functional implications of multiple isoforms. Physiol Rev. 1995;75:725-48. [ Links ]
13. Sunahara RK, Taussig R. Isoforms of mammalian adenylyl cyclase: Multiplicities of signaling. Mol Interv. 2002;2:168-84. [ Links ]
14. Dessauer CW. Adenylyl cyclase-A-kinase anchoring protein complexes: The next dimension in cAMP signaling. Mol Pharmacol. 2009;76:935-41. [ Links ]
15. Francis SH, Blount MA, Corbin JD. Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol Rev. 2011;91:651-90. [ Links ]
16. Rang HP, Dale MM, Ritter JM, and Moore PK. Pharmacology. 5th ed. Edinburgh, UK: Elsevier; 2003. [ Links ]
17. Borger P, Postma DS, Vellenga E, Kauffman HF. Regulation of asthma- related T-cell cytokines by the cyclic AMP-dependent signalling pathway. Clin Exp Allergy. 2000;30:920-6. [ Links ]
18. Kwok RP, Lundblad JR, Chrivia JC, Richards JP, Bachinger HP, Brennan RG, et al. Nuclear protein CBP is a coactivator for the transcription factor CREB. Nature. 1994;370:223-6. [ Links ]
19. Gupta A, Terhorst C. CD3 delta enhancer. CREB interferes with the function of a murine CD3-delta A binding factor (M delta AF). J Immunol. 1994;152:3895-903. [ Links ]
20. Chow CW, Davis RJ. Integration of calcium and cyclic AMP signaling pathways by 14-3-3. Mol Cell Biol. 2000;20:702-12. [ Links ]
21. Torgersen KM, Vang T, Abrahamsen H, Yaqub S, Tasken K. Molecular mechanisms for protein kinase A-mediated modulation of immune function. Cell Signal. 2002;14:1-9. [ Links ]
22. Staples KJ, Bergmann M, Tomita K, Houslay MD, McPhee I, Barnes PJ, et al. Adenosine 3',5'-cyclic monophosphate (cAMP)-dependent inhibition of IL-5 from human T lymphocytes is not mediated by the cAMP-dependent protein kinase A. J Immunol. 2001;167:2074-80. [ Links ]
23. Henning SW, Cantrell DA. GTPases in antigen receptor signalling. Curr Opin Immunol. 1998;10:322-9. [ Links ]
24. Seddiki N, Santner-Nanan B, Martinson J, Zaunders J, Sasson S, Landay A, et al. Expression of interleukin (IL)-2 and IL-7 receptors discriminates between human regulatory and activated T cells. J Exp Med. 2006;203:1693-700. [ Links ]
25. Workman CJ, Szymczak-Workman AL, Collison LW, Pillai MR, Vignali DA. The development and function of regulatory T cells. Cell Mol Life Sci. 2009;66:2603-22. [ Links ]
26. Placke T, Kopp HG, Salih HR. Glucocorticoid-induced TNFR-related (GITR) protein and its ligand in antitumor immunity: Functional role and therapeutic modulation. Clin Dev Immunol. 2010:239083 27. [ Links ] Huang B, Zhao J, Lei Z, Shen S, Li D, Shen GX, et al. miR-142-3p restricts cAMP production in CD4+CD25- T cells and CD4+CD25+ TREG cells by targeting AC9 mRNA. EMBO Rep. 2009;10:180-5. [ Links ]
28. Barthlott T, Moncrieffe H, Veldhoen M, Atkins CJ, Christensen J, O'Garra A, et al. CD25+ CD4+ T cells compete with naive CD4+ T cells for IL-2 and exploit it for the induction of IL-10 production. Int Immunol 2005;17:279-88. [ Links ]
29. Bazhin AV, Kahnert S, Kimpfler S, Schadendorf D, Umansky V. Distinct metabolism of cyclic adenosine monophosphate in regulatory and helper CD4+ T cells. Mol Immunol. 2010;47:678-84. [ Links ]
30. Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445:931-5. [ Links ]
31. Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445:936-40. [ Links ]
32. Johansson CC, Bryn T, Yndestad A, Eiken HG, Bjerkeli V, Froland SS, et al. Cytokine networks are pre-activated in T cells from HIV-infected patients on HAART and are under the control of cAMP. AIDS. 2004;18:171-9. [ Links ]
33. Bopp T, Dehzad N, Reuter S, Klein M, Ullrich N, Stassen M, et al. Inhibition of cAMP degradation improves regulatory T cell-mediated suppression. J Immunol. 2009;182:4017-24. [ Links ]
34. Yaqub S, Tasken K. Role for the cAMP-protein kinase A signaling pathway in suppression of antitumor immune responses by regulatory T cells. Crit Rev Oncog. 2008;14:57-77. [ Links ]
35. Vendetti S, Patrizio M, Riccomi A, De Magistris MT. Human CD4+ T lymphocytes with increased intracellular cAMP levels exert regulatory functions by releasing extracellular cAMP. J Leukoc Biol.. 2006;80:880-8. [ Links ]
36. Vendetti S, Riccomi A, Sacchi A, Gatta L, Pioli C, De Magistris MT. Cyclic adenosine 5'-monophosphate and calcium induce CD152 (CTLA-4) up-regulation in resting CD4+ T lymphocytes. J Immunol. 2002;169:6231-5. [ Links ]
37. Li K, Anderson KJ, Peng Q, Noble A, Lu B, Kelly AP, et al. Cyclic AMP plays a critical role in C3a-receptor-mediated regulation of dendritic cells in antigen uptake and T-cell stimulation. Blood. 2008;112:5084-94. [ Links ]
38. Kambayashi T, Wallin RP, Ljunggren HG. cAMP-elevating agents suppress dendritic cell function. J Leukoc Biol. 2001;70:903-10. [ Links ]
39. la Sala A, He J, Laricchia-Robbio L, Gorini S, Iwasaki A, Braun M, et al. Cholera toxin inhibits IL-12 production and CD8alpha+ dendritic cell differentiation by cAMP-mediated inhibition of IRF8 function. J Exp Med. 2009;206:1227-35. [ Links ]
40. Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin- 10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683-765. [ Links ]
41. Steinbrink K, Graulich E, Kubsch S, Knop J, Enk AH. CD4(+) and CD8(+) anergic T cells induced by interleukin-10-treated human dendritic cells display antigen-specific suppressor activity. Blood. 2002;99:2468-76. [ Links ]
42. Koppelman B, Neefjes JJ, de Vries JE, de Waal Malefyt R. Interleukin- 10 down-regulates MHC class II alphabeta peptide complexes at the plasma membrane of monocytes by affecting arrival and recycling. Immunity. 1997;7:861-71. [ Links ]
43. Platzer C, Fritsch E, Elsner T, Lehmann MH, Volk HD, Prosch S. Cyclic adenosine monophosphate-responsive elements are involved in the transcriptional activation of the human IL-10 gene in monocyticcells. Eur J Immunol. 1999;29:3098-104. [ Links ]
44. Zhou W, Hashimoto K, Goleniewska K, O'Neal JF, Ji S, Blackwell TS, et al. Prostaglandin I2 analogs inhibit proinflammatory cytokine production and T cell stimulatory function of dendritic cells. J Immunol. 2007;178:702-10. [ Links ]
45. Son Y, Ito T, Ozaki Y, Tanijiri T, Yokoi T, Nakamura K, et al. Prostaglandin E2 is a negative regulator on human plasmacytoid dendritic cells. Immunology. 2006;119:36-42. [ Links ]
46. Sakata D, Yao C, Narumiya S. Prostaglandin E2, an immunoactivator. J Pharmacol Sci. 2010;112:1-5. [ Links ]
47. Bryce PJ, Dascombe MJ, Hutchinson IV. Immunomodulatory effects of pharmacological elevation of cyclic AMP in T lymphocytes proceed via a protein kinase A independent mechanism. Immunopharmacology. 1999;41:139-46. [ Links ]
48. Fuld S, Borland G, Yarwood SJ. Elevation of cyclic AMP in Jurkat Tcells provokes distinct transcriptional responses through the protein kinase A (PKA) and exchange protein activated by cyclic AMP (EPAC) pathways. Exp Cell Res. 2005;309:161-73. [ Links ]
49. Boussiotis VA, Freeman GJ, Berezovskaya A, Barber DL, Nadler LM. Maintenance of human T cell anergy: Blocking of IL-2 gene transcription by activated Rap1. Science. 1997;278:124-8. [ Links ]
50. Garay J, D'Angelo JA, Park Y, Summa CM, Aiken ML, Morales E, et al. Crosstalk between PKA and Epac regulates the phenotypic maturation and function of human dendritic cells. J Immunol. 2010;185:3227-38. [ Links ]
51. Mandapathil M, Hilldorfer B, Szczepanski MJ, Czystowska M, Szajnik M, Ren J, et al. Generation and accumulation of immunosuppressive adenosine by human CD4+CD25highFOXP3+ regulatory T cells. J Biol Chem. 2010;285:7176-86. [ Links ]
52. Sitkovsky M, Lukashev D, Deaglio S, Dwyer K, Robson SC, Ohta A. Adenosine A2A receptor antagonists: Blockade of adenosinergic effects and T regulatory cells. Br J Pharmacol. 2008;153:S457-64. [ Links ]
53. Dong RP, Kameoka J, Hegen M, Tanaka T, Xu Y, Schlossman SF, et al. Characterization of adenosine deaminase binding to human CD26 on T cells and its biologic role in immune response.J Immunol. 1996;156:1349-55. [ Links ]
54. Majumdar S, Aggarwal BB. Adenosine suppresses activation of nuclear factor-kappaB selectively induced by tumor necrosis factor in different cell types.Oncogene. 2003;22:1206-18. [ Links ]
55. Haskó G, Kuhel DG, Németh ZH, Mabley JG, Stachlewitz RF, Virág L, et al. Inosine inhibits inflammatory cytokine production by a posttranscriptional mechanism and protects against endotoxin-induced shock. J Immunol. 2000;164:1013-9. [ Links ]
56. Sajjadi FG, Takabayashi K, Foster AC, Domingo RC, Firestein GS.Inhibition of TNF-alpha expression by adenosine: Role of A3 adenosine receptors. J Immunol. 1996;156:3435-42. [ Links ]
57. McWhinney CD, Dudley MW, Bowlin TL, Peet NP, Schook L, Bradshaw M, et al. Activation of adenosine A3 receptors on macrophages inhibits tumor necrosis factor-alpha. Eur J Pharmacol. 1996;310:209-16. [ Links ]
58. Jin X, Shepherd RK, Duling BR, Linden J. Inosine binds to A3 adenosine receptors and stimulates mast cell degranulation. J Clin Invest. 1997;100:2849-57. [ Links ]
59. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: Hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225-32. [ Links ]
60. Liao H, Hyman MC, Baek AE, Fukase K, Pinsky DJ. cAMP/CREBmediated transcriptional regulation of ectonucleoside triphosphate diphosphohydrolase 1 (CD39) expression. J Biol Chem. 2010;285:14791-805. [ Links ]
61. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257-65. [ Links ]
62. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T regulatory and primed uncommitted CD4 T cells express CD73, which suppresses effector CD4 T cells by converting 5'-adenosine monophosphate to adenosine. J Immunol. 2006;177:6780-6. [ Links ]
63. Erdmann AA, Gao ZG, Jung U, Foley J, Borenstein T, Jacobson KA, et al. Activation of Th1 and Tc1 cell adenosine A2A receptors directly inhibits IL-2 secretion in vitro and IL-2-driven expansion in vivo. Blood. 2005;105:4707-14. [ Links ]
64. Raskovalova T, Lokshin A, Huang X, Su Y, Mandic M, Zarour HM, et al. Inhibition of cytokine production and cytotoxic activity of human antimelanoma specific CD8+ and CD4+ T lymphocytes by adenosine- protein kinase A type I signaling. Cancer Res. 2007;67:5949-56. [ Links ]
65. Alam MS, Kurtz CC, Wilson JM, Burnette BR, Wiznerowicz EB, Ross WG, et al. A2A adenosine receptor (AR) activation inhibits pro-inflammatory cytokine production by human CD4+ helper T cells and regulates Helicobacter-induced gastritis and bacterial persistence. Mucosal Immunol. 2009;2:232-42. [ Links ]
66. Evans WH, Boitano S. Connexin mimetic peptides: Specific inhibitors of gap-junctional intercellular communication. Biochem Soc Trans. 2001;29:606-12. [ Links ]
67. Fonseca PC, Nihei OK, Savino W, Spray DC, Alves LA. Flow cytometry analysis of gap junction-mediated cell-cell communication: Advantages and pitfalls. Cytometry A. 2006;69:487-93. [ Links ]
68. Vaeth M, Gogishvili T, Bopp T, Klein M, Berberich-Siebelt F, Gattenloehner S, et al. Regulatory T cells facilitate the nuclear accumulation of inducible cAMP early repressor (ICER) and suppress nuclear factor of activated T cell c1 (NFATc1). Proc Natl Acad Sci U S A. 2011;108:2480-5. [ Links ]
69. Moreno-Fernández ME, Rueda CM, Rusie LK, Chougnet CA. Regulatory T cells control HIV replication in activated T cells through a cAMP-dependent mechanism. Blood. 2011;117:5372-80. [ Links ]
70. Ring S, Karakhanova S, Johnson T, Enk AH, Mahnke K. Gap junctions between regulatory T cells and dendritic cells prevent sensitization of CD8(+) T cells. J Allergy Clin Immunol. 2010;125:237-46. [ Links ]
71. Fassbender M, Gerlitzki B, Ullrich N, Lupp C, Klein M, Radsak MP, et al. Cyclic adenosine monophosphate and IL-10 coordinately contribute to nTreg cell-mediated suppression of dendritic cell activation. Cell Immunol. 2010;265:91-6. [ Links ]
72. Leal DB, Streher CA, Bertoncheli C de M, Carli LF, Leal CA, da Silva JE, et al. HIV infection is associated with increased NTPDase activity that correlates with CD39-positive lymphocytes. Biochim Biophys Acta. 2005;1746:129-34. [ Links ]
73. Nigam P, Velu V, Kannanganat S, Chennareddi L, Kwa S, Siddiqui M, et al. Expansion of FOXP3+ CD8 T cells with suppressive potential in colorectal mucosa following a pathogenic simian immunodeficiency virus infection correlates with diminished antiviral T cell response and viral control. J Immunol. 2010;184:1690-701. [ Links ]
74. Hofmann B, Nishanian P, Nguyen T, Insixiengmay P, Fahey JL. Human immunodeficiency virus proteins induce the inhibitory cAMP/ protein kinase A pathway in normal lymphocytes. Proc Natl Acad Sci U S A. 1993;90:6676-80. [ Links ]
75. Masci AM, Galgani M, Cassano S, De Simone S, Gallo A, De Rosa V, et al. HIV-1 gp120 induces anergy in naive T lymphocytes through CD4-independent protein kinase-A-mediated signaling. J Leukoc Biol. 2003;74:1117-24. [ Links ]
76. Becker C, Taube C, Bopp T, Michel K, Kubach J, Reuter S, et al. Protection from graft-versus-host disease by HIV-1 envelope protein gp120-mediated activation of human CD4+CD25+ regulatory T cells. Blood. 2009;114:1263-9. [ Links ]
77. Epple HJ, Loddenkemper C, Kunkel D, Troger H, Maul J, Moos V, et al. Mucosal but not peripheral FOXP3+ regulatory T cells are highly increased in untreated HIV infection and normalize after suppressive HAART. Blood. 2006;108:3072-8. [ Links ]
78. Martínez-Navio JM, Climent N, Pacheco R, García F, Plana M, Nomdedeu M, et al. Immunological dysfunction in HIV-1-infected individuals caused by impairment of adenosine deaminase-induced costimulation of T-cell activation. Immunology. 2009;128:393-404. [ Links ]
79. Valenzuela A, Blanco J, Callebaut C, Jacotot E, Lluis C, Hovanessian AG, et al. HIV-1 envelope gp120 and viral particles block adenosine deaminase binding to human CD26. Adv Exp Med Biol. 1997;421:185-92. [ Links ]
80. Blanco J, Valenzuela A, Herrera C, Lluis C, Hovanessian AG, Franco R. The HIV-1 gp120 inhibits the binding of adenosine deaminase to CD26 by a mechanism modulated by CD4 and CXCR4 expression. FEBS Lett. 2000;477:123-8. [ Links ]
81. Wrenger S, Reinhold D, Faust J, Mrestani-Klaus C, Brandt W, Fengler A, et al. Effects of nonapeptides derived from the N-terminal structure of human immunodeficiency virus-1 (HIV-1) Tat on suppression of CD26-dependent T cell growth. Adv Exp Med Biol. 2000;477:161-5. [ Links ]
82. Navarro J, Punzon C, Jiménez JL, Fernández-Cruz E, Pizarro A, Fresno M, et al. Inhibition of phosphodiesterase type IV suppresses human immunodeficiency virus type 1 replication and cytokine production in primary T cells: Involvement of NF-kappaB and NFAT. J Virol. 1998;72:4712-20. [ Links ]
83. Rincón M, Tugores A, López-Rivas A, Silva A, Alonso M, De Landazuri MO, et al. Prostaglandin E2 and the increase of intracelular cAMP inhibit the expression of interleukin 2 receptors in human T cells. Eur J Immunol. 1988;18:1791-6. [ Links ]
84. Hayes MM, Lane BR, King SR, Markovitz DM, Coffey MJ. Prostaglandin E(2) inhibits replication of HIV-1 in macrophages through activation of protein kinase A. Cell Immunol. 2002;215:61-71. [ Links ]
85. Thivierge M, Le Gouill C, Tremblay MJ, Stankova J, Rola-Pleszczynski M. Prostaglandin E2 induces resistance to human immunodeficiency virus-1 infection in monocyte-derived macrophages: Down-regulation of expression by cyclic adenosine monophosphate. Blood. 1998;92:40-5. [ Links ]
86. Barat C, Gilbert C, Imbeault M, Tremblay MJ. Extracellular ATP reduces HIV-1 transfer from immature dendritic cells to CD4+ T lymphocytes. Retrovirology. 2008;5:30. [ Links ]
87. By Y, Durand-Gorde JM, Condo J, Lejeune PJ, Fenouillet E, Guieu R, et al. Monoclonal antibody-assisted stimulation of adenosine A2A receptors induces simultaneous down-regulation of CXCR4 and CCR5 on CD4+ T-cells. Hum Immunol. 2010;71:1073-6. [ Links ]

