










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkInfectio
Print version ISSN 0123-9392
Infect. vol.20 no.1 Bogotá Jan./March 2016
https://doi.org/10.1016/j.infect.2015.07.002
REVISIÓN
http://dx.doi.org/10.1016/j.infect.2015.07.002
Abordaje inmunológico del síndrome por deleción 22q11Estefanía Vásquez-Echeverri a , Federico Sierra a , Claudia M. Trujillo-Vargas a , Julio C. Orrego-Arango a , Carlos Garcés-Samudio b , c , Rafael Lince c , d , e y José L. Franco-Restrepo a , *
a Grupo de Inmunodeficiencias Primarias, Universidad de Antioquia
b Grupo Pediaciencias, Universidad de Antioquia
c Hospital Pablo Tobón Uribe
d Clínica CardioVID
e Hospital Universitario San Vicente, Fundación Medellín, Antioquia, Colombia
Recibido el 25 de marzo de 2015; aceptado el 31 de julio de 2015
Disponible en Internet el 3 de noviembre de 2015
Resumen El síndrome por deleción 22q11 (SD22q11) es el síndrome por deleción cromosómica más frecuente en humanos y se caracteriza por la tríada clínica que incluye cardiopatía congénita, hipocalcemia e inmunodeficiencia primaria. El 85-90% de los pacientes tienen microdeleciones en el cromosoma 22q11.2. Tomando como punto cardinal la cardiopatía congénita, se diseñó una estrategia para tamización y diagnóstico de SD22q11 con énfasis en la evaluación inmune. Es imprescindible realizar una historia clínica detallada y, posteriormente, un análisis cuantitativo y funcional de las subpoblaciones de linfocitos en sangre periférica para clasificarlo en SD22q11 completo (<1%) o parcial (95-99%) e instaurar las pautas de tratamiento en aspectos como: aislamiento del paciente, vacunación, profilaxis contra microorganismos oportunistas, uso de productos sanguíneos irradiados y reconstitución inmunológica. Sin embargo, el abordaje del paciente debe ser multidisciplinario para detectar y prevenir complicaciones a largo plazo que pueden ser graves, especialmente en los pacientes con SD22q11 completo.
PALABRAS CLAVE
Síndrome de DiGeorge; Síndrome por deleción 22q11; Cardiopatía troncoconal; Inmunodeficiencia; Linfopenia congénita; Hipoplasia tímica; Aplasia tímica
© 2015 Publicado por Elsevier España, S.L.U. en nombre de ACIN. Este es un artículo Open Access bajo la licencia CC BY-NC-ND ( http://creativecommons.org/licenses/by-nc-nd/4.0/ ).
Immunological approaches to 22q11 deletion syndrome
Abstract In humans, 22q11 deletion syndrome (22q11DS) is considered the most common chromosome deletion syndrome. It is characterised by a clinical triad that includes congenital heart disease, hypocalcaemia and primary immunodeficiency. Approximately 85-90% of patients with this syndrome exhibit microdeletions in chromosome 22q11.2. Using congenital heart disease as a starting point, we designed a strategy for the screening and diagnosis of 22q11DS with an emphasis on immunological evaluation. A detailed clinical history and the subsequent quantitative and functional analyses of the lymphocyte subpopulations in the peripheral blood is crucial to classify as complete (<1%) or partial (95-99%) the disease and to guide clinicians in terms of patient isolation, vaccination, prophylaxis for opportunistic infections, use of irradiated blood products and immunological reconstitution. However, multidisciplinary care is necessary to detect and prevent long-term complications that could be severe, particularly in cases of complete 22q11DS.
KEYWORDS
DiGeorge syndrome; 22q11 deletion syndrome; Congenital heart disease; Immunodeficiency; Congenital lymphopaenia; Thymic hypoplasia; Thymic aplasia
© 2015 Published by Elsevier España, S.L.U. on behalf of ACIN. This is an open access article under the CC BY-NC-ND license ( http://creativecommons.org/licenses/by-nc-nd/4.0/ ).
http://dx.doi.org/10.1016/j.infect.2015.07.002
0123-9392/© 2015 Publicado por Elsevier España, S.L.U . en nombre de ACIN. Este es un artículo Open Access bajo la licencia CC BY-NC-ND ( http://creativecommons.org/licenses/by-nc-nd/4.0/ ).
Introducción
Angelo M. DiGeorge describió en 1965 un síndrome caracterizado por la ausencia congénita del timo y la glándula paratiroides, que tiempo después fue llamado síndrome de DiGeorge (SDG, OMIM #188400). Este trastorno se caracteriza clínicamente por la tríada clásica de cardiopatía congénita y endocrinopatía con hipocalcemia e inmunodeficiencia primaria debido a aplasia o hipoplasia de la paratiroides y el timo, respectivamente. No obstante, el síndrome puede exhibir múltiples anormalidades y manifestaciones clínicas pleiotrópicas que a menudo resultan en dismorfismo facial y alteraciones en el paladar, entre otros 1 . Ya que el 85-90% de los pacientes con SDG tienen microdeleciones hemicigóticas en el cromosoma 22q11.2 2, la nomenclatura actual lo ha redefinido como síndrome por deleción 22q11 (SD22q11) y así nos referiremos a este en el texto. El SD22q11 resulta de alteraciones en la migración de las células del neuroectodermo de la cresta neural, principalmente a la tercera y cuarta bolsas y arcos faríngeos, durante el desarrollo embrionario en humanos, aunque también existe evidencia clínica que n señala defectos del primer y sexto arcos y bolsas faríngeas 3 . Entre los genes asociados a defectos en la migración de las células derivadas de la cresta neural se encuentra el gen Tbx1 que está localizado en la región crítica del SD22q11. La haploinsuficiencia de este gen también ha sido relacionada con los defectos coronarios característicos del SD22q11 4 .
El SD22q11 es la deleción cromosómica más frecuente en humanos con una prevalencia estimada de 1 por cada 4.000 nacidos vivos, afecta por igual a ambos sexos y su relevancia clínica radica en que es la segunda causa de retraso del desarrollo y una de las principales causas de cardiopatía congénita después del síndrome de Down 5 .
Las anormalidades inmunes usualmente son el resultado de un desarrollo tímico deficiente. La aplasia es la alteración más severa, aunque se presenta en <1% de los casos (SD22q11 completo), mientras que la hipoplasia tímica se observa en más del 95% (SD22q11 parcial) 6 . A nivel celular, la disfunción de linfocitos T (LT) ya sea en número o función es la característica más frecuente ya que la mayoría de los pacientes tienen número normal de linfocitos B (LB) y NK; clínicamente se evidencian en mayor susceptibilidad a infecciones respiratorias o gastrointestinales recurrentes 7 . En los casos de aplasia tímica, se hace necesario el tratamiento con antibióticos profilácticos y trasplante tímico, mientras en los otros casos se hace manejo expectante 7.
Debido al desconocimiento por parte del personal médico del compromiso multisistémico de la enfermedad y, particularmente, de las anormalidades inmunes, la presente revisión está orientada a diseñar una estrategia para tamización, diagnóstico y manejo multidisciplinario de este síndrome con énfasis en el estudio de su función inmune, tomando como punto cardinal la cardiopatía congénita, con el fin de brindar un seguimiento adecuado para la detección temprana de complicaciones y de mejorar la calidad de vida tanto de los pacientes como de sus familias.
Características clínicas más relevantes en el SD22q11
La tabla 1 resume la frecuencia de las diferentes manifestaciones clínicas identificadas en el SD22q11. Es necesario resaltar que, debido a la expresividad variable del defecto genético en el síndrome, pueden presentarse pacientes con compromiso severo que lleva a muerte neonatal temprana, mientras en otros se presentan tan pocas características clínicas típicas del síndrome o se presentan con un compromiso tan bajo que pueden pasar desapercibidas y solo diagnosticarse durante la vida adulta 8.

Malformaciones cardiovasculares congénitas
Son una de las manifestaciones más comunes del SD22q11; las de tipo troncoconal son las más frecuentes ( tabla 1, fig. 1 ). Otras alteraciones incluyen atresia pulmonar, estenosis de la arteria subclavia, origen cervical de la arteria subclavia y transposición de grandes arterias 1 . En algunas ocasiones, los defectos troncoconales pueden ir acompañados de otros menos frecuentes como comunicación interauricular o la comunicación interventricular y los defectos valvulares como estenosis valvular pulmonar, entre otros 9 .
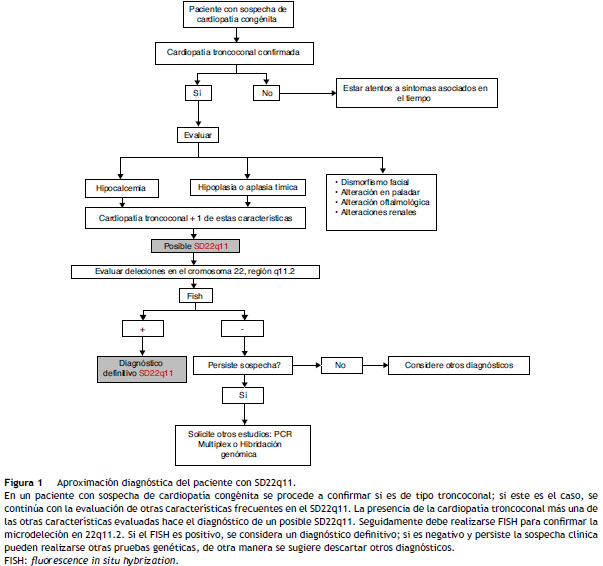
La evaluación inicial del paciente por un cardiólogo pediatra es fundamental para la identificación del tipo de cardiopatía. Aparte de los hallazgos clínicos que incluyen presencia de -cianosis, dificultades en la alimentación, fatiga, falla en el medro, entre otros, son fundamentales las ayudas diagnósticas como los rayos X de tórax, el electrocardiograma y la ecocardiografía. Según los resultados de esta evaluación clínica, el tipo de cardiopatía y el compromiso hemodinámico, se decidirá si se realizan intervenciones quirúrgicas o solo manejo expectante 10.
Hipocalcemia
Es causada por el hipoparatiroidismo que resulta de la hipoplasia de las glándulas paratiroideas y suele manifestarse de manera variable, particularmente durante el periodo neonatal. Existen 2 presentaciones de la hipocalcemia neonatal: la de inicio temprano que se presenta en las primeras 72 h de vida y es generalmente asintomática y la de inicio tardío, que se presenta generalmente luego del séptimo día de vida y puede acompañarse de irritabilidad, llanto de tono alto, temblores, convulsiones, apnea, cianosis, hipotonía, hipertonía, vómito y prolongación del intervalo QTc > 0,4 s 9,11 . En algunos casos la hipocalcemia puede manifestarse luego que el paciente es sometido a cirugía para la corrección de su cardiopatía congénita, debido a la demanda metabólica requerida durante el procedimiento quirúrgico ( fig. 1 ) 1 .
El diagnóstico se realiza por la disminución de los niveles séricos de calcio ionizado y hormona paratiroidea de acuerdo con los valores normales establecidos para la edad 5. El tratamiento de elección es la suplementación con calcio y vitamina D en la dieta, especialmente durante la adolescencia. Sin embargo, en la mayoría de los casos no es necesaria la suplementación permanente, ya que la hipocalcemia tiende a mejorar en el primer año de vida, por lo cual se requiere la evaluación por Endocrinología 1,5.
Hipoplasia o aplasia del timo
Se evidencia usualmente en las radiografías de tórax durante el estudio de la cardiopatía, y constituye una de las características principales, dando lugar a disfunción de LT. Las infecciones pueden presentarse antes, durante o después de la cirugía para la corrección del defecto cardíaco, con consecuencias significativas en cuanto a morbimortalidad. Por lo tanto, debe realizarse una evaluación inmunológica de rutina en todo paciente con cardiopatía congénita, particularmente en los pacientes afectados por las del tipo observado en SD22q11 ( fig. 1 ).
Otras manifestaciones
Otras manifestaciones que facilitan el reconocimiento de los pacientes son el hipertelorismo, pabellones auriculares prominentes con implantación baja y micrognatia. Otras menos comunes incluyen asimetría facial, retrognatia, epicanto, microtia o anotia, entre otras 8 . Algunas de estas características pueden hacerse más prominentes en la niñez y en la adolescencia. Adicionalmente, se debe estar atento a la aparición de otras manifestaciones clínicas que, aunque ocurren con menor frecuencia, también requieren de un manejo especializado ( tabla 1 ).
Diagnóstico molecular
El objetivo final de la evaluación del paciente con sospecha de SD22q11 es identificar las bases genéticas de la enfermedad, confirmando el diagnóstico clínico y de laboratorio ( fig. 1 ). Esto permite, además, conocer la epidemiología de la enfermedad y, de esta forma, favorecer la implementación de políticas públicas para establecer programas de tamización y facilitar los trámites administrativos en cuanto a la autorización del tratamiento dirigido y manejo integral de estos pacientes. Es importante realizar estos estudios también en los padres pues, aunque en el 90% de los pacientes con SD22q11 la presentación es esporádica, hasta en el 10% se demuestra herencia uniparental con transmisión autosómica dominante, lo cual implica que la enfermedad puede aparecer nuevamente en la descendencia y no ser apreciada en ciertos casos por compensación genética 12,13 .
La identificación del defecto genético puede hacerse mediante citogenética inicialmente con un cariotipo con bandeo G de alta resolución. Sin embargo, su positividad en la detección de microdeleciones suele ser menor al 25% 14 . Por lo tanto, es más conveniente realizar FISH empleando sondas específicas, pues este es considerado como el método estándar para la confirmación del diagnóstico por su sensibilidad que es >80% para la detección de las microdeleciones asociadas al SD22q11. En los casos en los cuales no se detectan deleciones en este cromosoma (alrededor del 5-10%), deben realizarse estudios adicionales como la PCR multiplex o la hibridación genómica comparativa 5,13 .
Diagnóstico diferencial
Debido al espectro de manifestaciones clínicas tan amplio del SD22q11, anteriormente en la literatura este se describía con diferentes nombres. Por consiguiente, muchos miembros de la comunidad médica creen erróneamente que cada uno representa un síndrome distinto. Entre ellos, los más reconocidos han sido el SDG, el síndrome de Takao, CATCH 22 y el síndrome velocardiofacial 2 . Este último, sin embargo, se caracteriza por insuficiencia velofaríngea, cardiopatías troncoconales y dismorfismo facial sin la deficiencia inmune 15. Otros diagnósticos diferenciales incluyen el cat eye syndrome causado por tetrasomia o trisomía 22q 16 .
Abordaje inmunológico inicial del paciente con sospecha de SD22q11
La evaluación del paciente con sospecha de SD22q11 por un inmunólogo clínico debe ser parte fundamental del abordaje integral del paciente. Para la evaluación inmune inicial del paciente con SD22q11 es necesaria una historia clínica detallada de los antecedentes personales, familiares, esquema y efectos adversos de la vacunación, así como una descripción de la frecuencia, localización, gravedad y microorganismos causantes de las infecciones ( fig. 2 ). Las infecciones se presentan principalmente en los tractos respiratorio superior e inferior (otitis media, sinusitis, neumonías) y el tracto gastrointestinal (diarrea crónica) y su gravedad se evalúa, además, por la necesidad de unidad de cuidados intensivos, antibióticos intravenosos, tratamientos o estancias hospitalarias prolongadas. También es muy importante determinar la sensibilidad antibiótica de los microorganismos aislados. Adicionalmente, cuando se sospeche un proceso infeccioso viral, es necesario realizar los estudios de antigenemia, títulos de anticuerpos específicos y pruebas de biología molecular disponibles (carga viral) y definir la etiología.

Los microorganismos que más frecuentemente causan las infecciones en el SD22q11 son los enterovirus, adenovirus, parainfluenza y rotavirus, aunque también se documentan ocasionalmente infecciones por citomegalovirus y virus de Epstein-Barr 1 . Otros menos comunes incluyen bacterias, micobacterias (infección posvacunal con M. bovis o BCG diseminadas), parásitos y hongos ( Candida spp., Aspergillus y Pneumocystis jirovecii ) 17,18. No obstante, estos últimos, considerados oportunistas, son más relevantes en el SD22q11 completo (inmunodeficiencia grave), en cuyo caso el cuadro clínico suele ser indistinguible del que se presenta en una inmunodeficiencia combinada severa 19 . Deben tenerse en cuenta otros factores de riesgo como el estrato socioeconómico y las condiciones ambientales que determinan la exposición y colonización microbiana, así como defectos anatómicos asociados y otros como alergias y desnutrición, que contribuyen a aumentar la frecuencia y gravedad de las infecciones 20-22.
Estudio del timo
Los estudios imaginológicos durante la evaluación del paciente con cardiopatía congénita se incluyen dentro del abordaje inicial de pacientes con sospecha de SD22q11, ya que es una ayuda diagnóstica común, rápida y económica de evaluar la sombra tímica ( fig. 2 ). Debe tenerse en cuenta que el timo es sensible a los rayos X y se reduce de tamaño rápidamente luego de una radiografía simple o incluso por una sepsis, debido a que el estrés metabólico promueve la involución tímica, por lo cual la radiografía inicial es determinante 23. Adicionalmente, en algunos casos podría ser útil realizar ecografía, tomografía axial computarizada o resonancia nuclear magnética, particularmente en aquellos pacientes en los que se sospeche tejido tímico accesorio o de localización aberrante, aunque algunos proponen que en estos casos deben emplearse adicionalmente biomarcadores como los T cell Receptor Excision Circles (TREC) junto con pruebas funcionales 24 .
Evaluación cuantitativa de los linfocitos en sangre periférica
Hemoleucograma
Como una aproximación inicial, el hemoleucograma seriado permite determinar si existe linfopenia y si esta es persistente en el tiempo. Esta prueba debe valorarse según los parámetros de normalidad para la edad, teniendo en cuenta que valores absolutos de linfocitos totales disminuidos indican una alteración en los LT, ya que entre el 50 y el 70% de los linfocitos totales circulantes son LT 25. Este hallazgo, sin embargo, debe siempre ser confirmado mediante pruebas más especializadas para evaluar diferencialmente las subpoblaciones de linfocitos en sangre periférica.
Subpoblaciones de linfocitos en sangre periférica
La cuantificación de los LT, LB, NK y monocitos en sangre periférica mediante citometría de flujo constituye un análisis fundamental para caracterizar la linfopenia en los pacientes con SD22q11 ( fig. 2 ) 26, ya que algunos pacientes presentan disminución de LB particularmente en la infancia, pero estos se normalizan con el tiempo 27 . Por otra parte, al igual que los LT CD3+, los linfocitos NK disminuyen significativamente con la edad en los pacientes con SD22q11, aunque en algunos casos, pueden mantenerse estables y la relevancia clínica de este hallazgo es incierta 27 .
Si no se dispone de lo necesario para cuantificar los LT, LB, NK y monocitos en sangre periférica, un abordaje parcial para distinguir el tipo de linfopenia puede ser el « tritest » 28,29 . Esta prueba hace un recuento total de LT CD3+ y, adicionalmente, permite la cuantificación de aquellos LT CD3+ que son CD4+ o CD8+ calculando la relación CD4:CD8, aunque esta suele ser normal en la mayoría de los pacientes 27 .
Debe tenerse en cuenta que la inmunodeficiencia de LT en SD22q11 está relacionada con la edad, y es más marcada en el período neonatal y en el primer año de vida. Además, los números absolutos de estas células disminuyen de manera fisiológica a partir de los 2 años de edad, aproximadamente, pero este fenómeno sucede a un ritmo más lento que el normal en los pacientes con SD22q11 con compromiso inmune 27 . Por estas razones, esta evaluación debe realizarse al menos cada 6 meses hasta que los linfocitos alcancen los rangos de normalidad de acuerdo con la edad ( tabla 2 ).

Evaluación cualitativa de LT
Respuesta proliferativa a mitógenos
Ya que la disfunción inmune en el SD22q11 puede manifestarse con o sin disminución en los recuentos de LT CD3+ en sangre periférica 17,30, además de las pruebas cuantitativas, debe realizarse una prueba de proliferación en respuesta a mitógenos como la fitohemaglutinina para la evaluación funcional de los LT ( fig. 2 ). Esta debe hacerse al momento del diagnóstico y repetirse al menos de 6 meses a un año luego de realizada la valoración inicial para estudiar la evolución de la linfopenia ( tabla 2 ) 29,31 . La mayoría de los pacientes con SD22q11 exhiben linfoproliferación normal. Sin embargo, en algunos se observará disminuida, pero usualmente se normaliza en casi todos hacia los 3 años de edad. Algunos autores consideran esta prueba como el parámetro más importante para evaluar la función de LT y caracterizar el compromiso inmune en estos pacientes 31 .
Como se muestra en el figura 2, la evaluación inmunológica inicial permite subdividir el paciente con SD22q11 en:
SD22q11 completo con inmunodeficiencia grave: se define por recuentos absolutos de LT CD3+ <500 células/mL con respuestas linfoproliferativas muy disminuidas o ausentes (http://esid.org/Working-Parties/Clinical/Resources/Diagnostic-criteria-for-PID2#Q5) 5 . Sin embargo, el SD22q11 « completo » puede ser « atípico » cuando, además de las infecciones recurrentes de moderadas a severas, se detectan otras manifestaciones clínicas que incluyen rash generalizado (que puede ser de leve a moderado) acompañado de linfadenopatías y, ocasionalmente, hepatoesplenomegalia. En estos pacientes, la linfopenia suele no ser tan severa ( ~ 10%), y esto se explica por la presencia de algunos LT altamente autorreactivos, los cuales proliferan rápidamente después del nacimiento, enmascarando la linfopenia y causando manifestaciones autoinmunes 32 . Los pacientes con inmunodeficencia grave usualmente empeoran con el tiempo y requieren rápidamente un trasplante de explantes tímicos o, alternativamente, de médula ósea como tratamiento definitivo 3.
SD22q11 parcial con inmunodeficiencia leve a moderada: se define por valores de LT CD3+ en sangre periférica entre 500 y 1.500 células/mL con respuestas linfoproliferativas levemente disminuidas. Ocasionalmente puede resultar en infecciones pero suele tener un buen pronóstico a largo plazo, lo que indica, en la mayoría de los casos, un manejo expectante ya que la función y número de LT tiende a normalizarse con el tiempo 33 .
Seguimiento inmunológico del paciente con SD22q11
Posteriormente a la evaluación inicial de cada paciente, se debe realizar un plan de seguimiento riguroso para determinar la evolución de la inmunodeficiencia en cuanto a su resolución o persistencia ( tabla 2 ). En algunos pacientes se identifican anormalidades inmunológicas adicionales. Sin embargo, la madurez inmunológica que se adquiere con la edad es importante antes de considerar algunas de las pruebas que detallamos a continuación 14 .
Respuesta proliferativa de LT a antígenos específicos
Debe evaluarse a partir del año de vida, ya que el paciente debe haber sido inmunizado previamente 24 . Algunos consideran que esta prueba es más sensible para detectar disfunción inmune en los pacientes y correlaciona mejor con algunas otras pruebas, por lo cual es recomendable realizarla de manera simultánea con los de respuesta proliferativa a mitógenos 24 .
Dosificación de inmunoglobulinas séricas
Las anormalidades en el número o función de LT en SD22q11 puede afectar la producción de ciertos isotipos de inmunoglobulinas (Ig) e incluso los títulos protectores de anticuerpos específicos en respuesta a la vacunación, contribuyendo a aumentar la susceptibilidad de infecciones recurrentes. En algunos pacientes con SD22q11 puede observarse deficiencia de IgG, IgM, IgA o subclases de IgG 34 . También se puede observar en algunos casos hipergammaglobulinemia, aunque el significado de este hallazgo es desconocido 35 . Por consiguiente, se debe realizar dosificación inicial de IgM, IgG, IgA e IgE en el suero y comparar los valores de acuerdo con la edad y sexo del paciente 7,36.
En algunos casos es necesaria una electroforesis de proteínas para confirmar la hipogammaglobulinemia cuando la dosificación de Ig séricas no es concluyente. En esta última prueba se define hipogammaglobulinemia con un valor de la fracción gamma de las proteínas séricas <500 mg/dL, y agammaglobulinemia <200 mg/dL. Sin embargo se deben tener en cuenta los rangos de normalidad establecidos para la edad 37 .
Es necesario realizar monitoración de Ig séricas al menos después del primer año de edad y luego cada 6 a 12 meses, dependiendo de la evolución clínica, para determinar la necesidad de terapia de reemplazo de Ig 7,14,36.
Cuantificación de anticuerpos específicos en respuesta a la vacunación
En algunos pacientes puede observarse susceptibilidad elevada a infecciones recurrentes aún cuando los recuentos de LT e Ig séricas sean normales ya que puede haber una alteración en la producción de anticuerpos específicos 7,38. Por lo tanto, es necesario evaluar en edad preescolar también la producción de IgG en respuesta a antígenos T dependientes, tales como tétanos, difteria, virus de hepatitis B o rubéola, esta última, solo en los casos en que es permitido inmunizar con vacunas replicativas. Adicionalmente, para antígenos T-independientes se evalúan anticuerpos IgG contra varios de los serotipos contenidos en las vacunas disponibles para neumococo 38,39.
Pautas de manejo clínico de la inmunodeficiencia en pacientes con SD22q11
A continuación se exponen las recomendaciones claves para el manejo de la inmunodeficiencia en pacientes con SD22q11, que deben ser individualizadas según su compromiso inmune (de leve a moderado o grave) y condición clínica.
Aislamiento del paciente
El aislamiento del paciente es necesario si se documenta inmunodeficiencia grave hasta que pueda realizarse la reconstitución inmune (ver tabla 3 ). Esto implica principalmente aislamiento de contacto y respiratorio a nivel hospitalario. En casa, se debe evitar el contacto con personas con enfermedades infectocontagiosas y se debe insistir en la higiene para la preparación de los alimentos y el lavado de manos.

Vacunación
Hasta el momento, no existen guías publicadas para la administración de vacunas en pacientes con SD22q11, pero la evidencia clínica disponible señala que la administración de vacunas replicativas como la varicela y triple viral es segura en pacientes con compromiso de leve a moderado de la inmunidad y recuentos de LT CD4+ > 400 células/mL 40,41 . Se han reportado efectos adversos leves como rash, inflamación local, fiebre y linfadenopatías, pero no ha sido posible definir sus factores predictores 40,42 . Azzari et al. evaluaron la respuesta inmune y los efectos adversos a la vacuna contra el sarampión en 14 pacientes con SD22q11 con compromiso inmune de leve a moderado. Se observaron títulos protectores de anticuerpos contra el virus y efectos adversos leves que no fueron diferentes de los observados en la población general 41 .
La situación es diferente en pacientes con inmunodeficiencia grave, en los cuales están contraindicadas las vacunas replicativas hasta que se restablezca la función inmune mediante trasplante 14,40-43 .
Profilaxis contra microorganismos oportunistas
Para prevenir enfermedades por microorganismos oportunistas, principalmente Pneumocystis jirovecii , cuando el paciente presente inmunodeficiencia grave, se recomienda profilaxis con trimetropin sulfametoxazol (TMP-SMX) 7. Adicionalmente, está recomendada la profilaxis con fluconazol para prevenir candidiasis mucocutánea y algunos autores consideran el uso de aciclovir para prevenir infecciones por herpes virus 44,45 . Debe tenerse en cuenta que la profilaxis antimicrobiana se realiza mientras persista la disfunción inmune; sin embargo, el objetivo fundamental en el paciente con SD22q11 completo es la reconstitución inmunológica por medio de un trasplante. En la tabla 4 se incluyen los esquemas actualmente disponibles de profilaxis antimicrobiana.

Terapia de reemplazo con Ig humana
El objetivo del reemplazo con Ig es disminuir la frecuencia pero, sobre todo, la gravedad de las infecciones 36,46 . En un estudio realizado por Patel et al. que incluyó a 1.023 pacientes con SD22q11 en el cual se evaluaron los niveles de Ig, se reportó que solo 2-3% de ellos requieren la terapia de reemplazo 34 .
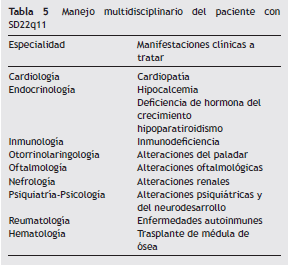
Independiente del tipo de inmunodeficiencia que se esté manejando, ESID establece que se debe considerar reemplazo con Ig en las siguientes situaciones: IgG < 200 mg/dl, excluyendo la hipogammaglobulinemia transitoria de la infancia sin infecciones recurrentes, IgG entre 200 y 500 mg/dL con inmunodeficiencia identificada y asociada a infecciones recurrentes e IgG > 500 mg/dL acompañada de deficiencia en la producción de anticuerpos específicos e infecciones recurrentes y graves. Previamente al inicio de la terapia, se deben solicitar los siguientes paraclínicos: IgG, IgM, IgA e IgE séricas, títulos de anticuerpos específicos a la vacunación contra neumococo, tétanos y difteria, hemoleucograma, subpoblaciones de linfocitos en sangre periférica, función hepática y renal 36 . En general, la dosis inicial recomendada de Ig para pacientes con hipogammaglobulinemia es de 400-600 mg/kg cada 3 a 4 semanas, aunque la terapia se debe individualizar de acuerdo con la gravedad de la hipogammaglobulinemia y las infecciones 46. El seguimiento de los niveles de IgG sérica se recomienda realizarlo en intervalos de 3 a 4 meses 36 ( tabla 5 ).
Uso de productos sanguíneos irradiados
Sin importar la gravedad de la inmunodeficiencia o si esta se ha documentado previamente o no, todos los pacientes que sean tratados mediante corrección quirúrgica de su cardiopatía congénita deben recibir productos sanguíneos irradiados con rayos gamma (efectivo para la prevención de la enfermedad injerto contra hospedero), desleucocitados y seronegativos para el citomegalovirus 7,47 .
Reconstitución inmunológica
El tratamiento óptimo para la deficiencia inmune en pacientes con SD22q11 completo es la reconstitución inmune mediante trasplante de explantes tímicos o de células madre para restaurar la función inmunológica 48.
Trasplante de timo
El trasplante de timo consiste en obtener tejido tímico de un donante, cultivarlo en el laboratorio para generar explantes y luego implantarlo ectópicamente en el músculo cuádriceps 49 . Aunque la histocompatibilidad HLA es deseable, no es requisito indispensable 50 . La reconstitución inmune se observa cuando se detectan LT funcionales, aproximadamente a los 3 o 6 meses luego del trasplante, y es frecuente observar más LT CD4+ que CD8+ 48 . También suele observarse la reconstitución de un repertorio de LT que es policlonal, lo cual indica que el injerto de timo tiene la capacidad de sostener el desarrollo de LT normales 9 .
Para la realización del trasplante de timo es necesario tener en cuenta que el receptor puede poseer algunos LT oligoclonales, los cuales pueden ser altamente autorreactivos y llevar al rechazo, por lo tanto, se debe estudiar la posibilidad de suministrar un régimen de inmunosupresión que incluya ciclosporina pre- y postrasplante y globulina antitimocito de conejo pretrasplante 49 .
Para evaluar la recuperación de la función tímica postrasplante, es útil cuantificar los emigrantes tímicos recientes mediante los TREC o por la expresión de los marcadores CD31 y CD45RA en LT CD4+ en sangre periférica 24,51,52 .
Trasplante de médula ósea
La transferencia adoptiva de células T maduras a través de un trasplante de médula ósea ha surgido como una alternativa para el SD22q11 completo cuando el trasplante de timo no está disponible 53 . Comparado con el trasplante de timo, este es un procedimiento fácil de realizar y se encuentra disponible en más centros de atención médica. Durante el seguimiento de un trasplante en pacientes con inmunodeficiencia, se debe ampliar la caracterización de las subpoblaciones de LT CD3 + CD4+ y CD3 + CD8+ mediante la utilización de marcadores de superficie como CD45RA, CD45RO y CCR7 52 .
Abordaje de las manifestaciones autoinmunes
La frecuencia de enfermedades autoinmunes es estimada entre el 10 y el 20% de los pacientes con SD22q11 y podrían ser consideradas como complicaciones a largo plazo. Las manifestaciones autoinmunes se asocian con oligoclonalidad de LT; sin embargo, no parecen relacionarse con el número de infecciones en estos pacientes 54 .
Entre las manifestaciones autoinmunes más frecuentemente descritas en SD22q11 se encuentran las citopenias, incluyendo anemia hemolítica y trombocitopenia autoinmune, artritis reumatoide, entre otros 8 .
Hasta el momento no hay biomarcadores que nos permitan identificar o predecir cuáles pacientes desarrollarán estas anormalidades autoinmunes; sin embargo, se propone, en caso de presentar sintomatología, realizar un hemoleucograma y evaluar autoanticuerpos, función tiroidea, antiglobulinas y, según los resultados de estas evaluaciones, hacer tratamiento y seguimiento respectivo guiado por el Servicio de Reumatología 7 .
Conclusiones
Esta estrategia constituye una recopilación de la experiencia clínica de los autores y de la evidencia científica disponible tanto a nivel nacional como internacional en el SD22q11 que pretende orientar a la comunidad médica acerca de la apropiada identificación, tamización, diagnóstico y seguimiento de estos pacientes.
Aunque la mayor parte de los casos registrados de SD22q11 presentan un fenotipo parcial con compromiso inmunológico leve, con pronóstico favorable a largo plazo, es necesario realizar un seguimiento de la función inmune para prevenir complicaciones. No obstante, la caracterización inmunológica es fundamental en los pacientes con fenotipo completo con compromiso severo de la función inmune en quienes la no vacunación con vacunas replicativas, la profilaxis contra microorganismos oportunistas, el uso de productos sanguíneos irradiados y desleucocitados y la reconstitución inmunológica temprana que restablece la función inmune de forma definitiva, radicalmente cambia el desenlace de la enfermedad y mejora el pronóstico a largo plazo.
Se consideran necesarios trabajos adicionales que hagan énfasis en otros aspectos clínicos que puedan estar afectados en estos pacientes para ofrecer las mejores alternativas, tanto diagnósticas como terapéuticas, buscando mejorar la calidad de vida y prevenir o tratar las posibles complicaciones no inmunes de forma oportuna, que también son muy frecuentes en SD22q11.
Responsabilidades éticas
Confidencialidad de los datos. Los autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Financiación
Este trabajo fue financiado por la estrategia de sostenibilidad 2011-2012 de la Universidad de Antioquia.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
* Autor para correspondencia.
Correo electrónico: jlfrancor@une.net.co (J.L. Franco-Restrepo).
Bibliografía
1. McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine (Baltimore). 2011;90:1-18. [ Links ]
2. Hacihamdioglu B, Hacihamdioglu D, Delil K. 22q11 deletion syndrome: Current perspective. Appl Clin Genet. 2015;8:123-32. [ Links ]
3. Graham Davies E. Immunodeficiency in DiGeorge syndrome and options for treating cases with complete athymia. Front Immunol. 2013;4:322. [ Links ]
4. Gao S, Li X, Amendt BA. Understanding the role of Tbx1 as a candidate gene for 22q11.2 deletion syndrome. Curr Allergy Asthma Rep. 2013;13:613-21. [ Links ]
5. Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr. 2011;159:332-9. [ Links ]
6. Markert ML, Devlin BH, Chinn IK, McCarthy EA. Thymus transplantation in complete DiGeorge anomaly. Immunol Res. 2009;44:61-70. [ Links ]
7. Gennery AR. Immunological aspects of 22q11.2 deletion syndrome. Cell Mol Life Sci. 2012;69:17-27. [ Links ]
8. Bales AM, Zaleski CA, McPherson EW. Newborn screening programs: Should 22q11 deletion syndrome be added? Genet Med. 2010;12:135-44. [ Links ]
9. Sullivan KE. Chromosome 22q11.2 deletion syndrome: DiGeorge syndrome-velocardiofacial syndrome. Inmunol Allergy Clin North Am. 2008;28:353-66. [ Links ]
10. Yeoh TY, Scavonetto F, Hamlin RJ, Burkhart HM, Sprung J, Weingarten TN. Perioperative management of patients with DiGeorge syndrome undergoing cardiac surgery. J Cardiothorac Vasc Anesth. 2014;28:995-1001. [ Links ]
11. Philip N, Reynaud R. Hypocalcemia and microdeletion 22q11.2. Arch Pediatr. 2008;15:648-9. [ Links ]
12. Carelle-Calmels N, Saugier-Veber P, Girard-Lemaire F, Rudolf G, Doray B, Guérin E, et al. Genetic compensation in a human genomic disorder. N Engl J Med. 2009;360:1211-6. [ Links ]
13. Habel A, Herriot R, Kumararatne D, Allgrove J, Baker K, Baxendale H, et al. Towards a safety net for management of 22q11.2 deletion syndrome: Guidelines for our times. Eur J Pediatr. 2014;173:757-65. [ Links ]
14. Driscoll DA, Sullivan KE. DiGeorge syndrome: A chromosome 22q11. 2 deletion syndrome. En: Ochs HD, Edvard Smith CI, Puck JM, editores. Primary inmunodeficiency diseases: A molecular and genetic approach. 2nd ed. New York: Oxford University Press; 2007. p. 485-93. [ Links ]
15. Ruda JM, Krakovitz P, Rose AS. A review of the evaluation and management of velopharyngeal insufficiency in children. Otolaryngol Clin North Am. 2012;45:653-69. [ Links ]
16. Chen Ch P, Ko T-M, Chen Y-Y, Su J-W, Wang W. Prenatal diagnosis and molecular cytogenetic characterization of mosaicism for a small supernumerary marker chromosome derived from chromosome 22 associated with cat eye syndrome. Gene. 2013;527:384-8. [ Links ]
17. Sullivan KE, Jawad AF, Randall P, Driscoll DA, Emanuel BS, McDonald-McGinn DM, et al. Lack of correlation between impaired T cell production, immunodeficiency, and other phenotypic features in chromosome 22q11. 2 deletion syndromes. Clin Immunol Immunopathol. 1998;86:141-6. [ Links ]
18. Chapel HM, Misbah S, Webster ADB. Assessment of the immune system. En: Ochs HD, Edvard Smith CI, Puck JM, editores. Primary inmunodeficiency diseases: A molecular and genetic approach. 2nd ed. New York: Oxford University Press.; 2007. p. 611-32. [ Links ]
19. Buckley RH. Pulmonary complications of primary immunodeficiencies. Paediatr Respir Rev. 2004;5 Suppl A:S225-33. [ Links ]
20. Bresnahan KA, Tanumihardjo SA. Undernutrition, the acute phase response to infection, and its effects on micronutrient status indicators. Adv Nutr. 2014;5:702-11. [ Links ]
21. Munyaka PM, Khafipour E, Ghia JE. External influence of early childhood establishment of gut microbiota and subsequent health implications. Front Pediatr. 2014;2:1-9. [ Links ]
22. Cocco JF, Antonetti JW, Burns JL, Heggers JP, Blackwell SJ. Characterization of the nasal, sublingual, and oropharyngeal mucosa microbiota in cleft lip and palate individuals before and after surgical repair. Cleft Palate Craniofac J. 2010;47: 151-5. [ Links ]
23. Nickels AS, Boyce T, Joshi A, Hagan J. Absence of the thymic shadow in a neonate suspected of primary immunodeficiency: Not a straightforward clinical sign of immunodeficiency. J Pediatr. 2015;166:203. [ Links ]
24. Lavi RF, Kamchaisatian W, Sleasman JW, Martin DP, Haraguchi S, Day NK. Thymic output markers indicate immune dysfunction in DiGeorge syndrome. J Allergy Clin Immunol. 2006;118: 1184-6. [ Links ]
25. Schatorjé EJ, Gemen EF, Driessen GJ, Leuvenink J, van Hout RW, de Vries E. Pediatric reference values for the peripheral T-cell compartment. Scand J Immunol. 2012;75:436-44. [ Links ]
26. Boldt A, Borte S, Fricke S, Kentouche K, Emmrich F, Borte M, et al. Eight-color immunophenotyping of T-, B-, and NK-cell subpopulations for characterization of chronic immunodeficiencies. Cytometry B Clin Cytom. 2014;86:191-206. [ Links ]
27. McLean-Tooke A, Barge D, Spickett GP, Gennery AR. Immunologic defects in 22q11.2 deletion syndrome. J Allergy Clin Immunol. 2008;122:362-7. [ Links ]
28. Sullivan KE, McDonald-McGinn D, Driscoll DA, Emanuel BS, Zackai EH, Jawad AF. Longitudinal analysis of lymphocyte function and numbers in the first year of life in chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Clin Diagn Lab Immunol. 1999;6:906-11. [ Links ]
29. Chinen J, Rosenblatt HM, Smith EO, Shearer WT, Noroski LM. Long-term assessment of T-cell populations in DiGeorge syndrome. J Allergy Clin Immunol. 2003;111:573-9. [ Links ]
30. Aglony M, Lizama M, Méndez C, Navarrete C, Garay F, Repetto G, et al. Clinical findings and immunologic variability in 9 patients with DiGeorge syndrome. Rev Med Chil. 2004;132:26-32 [ [ Links ]en español].
31. Yaganeh M, Gambineri E, Abolmaali K, Tamizifar B, Español T. Other well-defined immunodeficiencies. En: Rezaei N, Aghamohammadi A, Notarangelo LD, editores. Primary immunodeficiency diseases, definition, diagnosis and management. Berlín: Springer-Verlag; 2008. p. 251. [ Links ]
32. Hudson LL, Louise Markert M, Devlin BH, Haynes BF, Sempowski GD. Human T cell reconstitution in DiGeorge syndrome and HIV-1 infection. Semin Immunol. 2007;19:297-309. [ Links ]
33. Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Clin Immunol. 1999;93:190-9. [ Links ]
34. Patel K, Akhter J, Kobrynski L, Benjamin Gathmann MA, Davis O, Sullivan KE, et al. Immunoglobulin deficiencies: The B-lymphocyte side of DiGeorge syndrome. J Pediatr. 2012;161:950-3. [ Links ]
35. Gennery AR, Barge D, O'Sullivan JJ, Flood TJ, Abinun M, Cant AJ. Antibody eficiency and autoimmunity in 22q11.2 deletion syndrome. Arch Dis Child. 2002;86:422-5. [ Links ]
36. Condino-Neto A, Costa-Carvalho BT, Grumach AS, King A, Bezrodnik L, Oleastro M, et al. Guidelines for the use of human immunoglobulin therapy in patients with primary immunodeficiencies in Latin America. Allergol Immunopathol. 2014;42:245-60. [ Links ]
37. García D, Posada LH, Garcia LF, Cardona R. Niveles de inmunoglobulinas séricas en la población normal de Medellín. Acta Med Col. 1984;9:45-7. [ Links ]
38. Finocchi A, Di Cesare S, Romiti ML, Capponi C, Rossi P, Carsetti R, et al. Humoral immune responses and CD27+ B cells in children with DiGeorge syndrome (22q11.2 deletion syndrome). Pediatr Allergy Immunol. 2006;17:382-8. [ Links ]
39. Leal-Esteban LC, Rojas JL, Jaimes AL, Montoya JD, Montoya NE, Leiva L, et al. An immunoenzymatic test for IgG antibody levels against 10 serotypes of Streptococcus pneumoniae. Biomedica. 2012;32:92-102 [ [ Links ]en español].
40. Perez EE, Bokszczanin A, McDonald-McGinn D, Zackai EH, Sullivan KE. Safety of live viral vaccines in patients with chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/ velocardiofacial syndrome). Pediatrics. 2003;112:e325. [ Links ]
41. Azzari C, Gambineri E, Resti M, Moriondo M, Betti L, Saldias LR, et al. Safety and immunogenicity of measles-mumps-rubella vaccine in children with congenital immunodeficiency (DiGeorge syndrome). Vaccine. 2005;23:1668-71. [ Links ]
42. Al-Sukaiti N, Reid B, Lavi S, Al-Zaharani D, Atkinson A, Roifman CM, et al. Safety and efficacy of measles, mumps, and rubella vaccine in patients with DiGeorge syndrome. J Allergy Clin Immunol. 2010;126:868-9. [ Links ]
43. Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: The primary immune deficiency treatment consortium experience. J Allergy Clin Immunol. 2014;133:1092-8. [ Links ]
44. Freeman AF, Holland SM. Antimicrobial prophylaxis for primary immunodeficiencies. Curr Opin Allergy Clin Immunol. 2009;9:525-30. [ Links ]
45. Papadopoulou-Alakati E, Hassan A, Davies EG. Prevention of infection in c hildren and adolescents with primary immunodeficiency disorders. Asian Pac J Allergy Immunol. 2012;30: 249-58. [ Links ]
46. Bonagura VR. Illustrative cases on individualizing immunoglobulin therapy in primary immunodeficiency disease. Ann Allergy Asthma Immunol. 2013;111:S10-3. [ Links ]
47. Jatana V, Gillis J, Webster BH, Adès LC. Deletion 22q11.2 syndrome-implications for the intensive care physician. Pediatr Crit Care Med. 2007;8:459-63. [ Links ]
48. Markert ML, Devlin BH, Alexieff MJ, Li J, McCarthy EA, Gupton SE, et al. Review of 54 patients with complete DiGeorge anomaly enrolled in protocols for thymus transplantation: Outcome of 44 consecutive transplants. Blood. 2007;109:4539-47. [ Links ]
49. Markert ML, Devlin BH, McCarthy EA. Thymus transplantation. Clin Immunol. 2010;135:236-46. [ Links ]
50. Markert ML, Devlin BH, Chinn IK, McCarthy EA, Li YJ. Factors affecting success of thymus transplantation for complete DiGeorge anomaly. Am J Transplant. 2008;8:1729-36. [ Links ]
51. Kong F, Chen CH, Cooper MD. Thymic function can be accurately monitored by the level of recent T cell emigrants in the circulation. Immunity. 1998;8:97-104. [ Links ]
52. Toubert A, Glauzy S, Douay C, Clave E. Thymus and immune reconstitution after allogeneic hematopoietic stem cell transplantation in humans: Never say never again. Tissue Antigens. 2012;79:83-9. [ Links ]
53. McGhee SA, Lloret MG, Stiehm ER. Immunologic reconstitution in 22q deletion (DiGeorge) syndrome. Immunol Res. 2009;45:37-45. [ Links ]
54. McLean-Tooke A, Spickett GP, Gennery AR. Immunodeficiency and autoimmunity in 22q11. 2 deletion syndrome. Scand J Immunol. 2007;66:1-7. [ Links ]
