











 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
El virus de la hepatitis C (VHC) fue caracterizado molecularmente en 1989, reconociéndose como la causa mayor de hepatitis no-A, no-B (NANB) y una causa importante de las hepatitis crónicas a nivel mundial1,2. Durante este mismo año se desarrolló el primer enzimoinmunoensayo (ELISA) cuyo uso demostró que el VHC constituía la principal causa de hepatitis NANB post-transfusional en todo el mundo, el mayor responsable de la hepatitis NANB esporádica3.
Clasificación taxonómica
El VHC pertenece al género Hepacivirus, dentro de la familia Flaviviridae. Comparte esta familia con miembros del género Flavivirus (Por Ej. Virus del dengue, virus de la fiebre amarilla y virus del Nilo occidental) y con miembros del género Pestivirus (Por Ej. Virus de la diarrea bovina, virus del cólera porcino)4,5.
Características estructurales
Mediante microscopía crio-electrónica se ha demostrado que los viriones del VHC tienen una forma esférica con proyecciones en puntas y son heterogéneas de tamaño, desde 40 hasta 100nm de diámetro6. Mediante el análisis de partículas recombinantes del VHC con anticuerpos contra glicoproteínas, se ha identificado una envoltura externa, que consta de una bicapa lipídica donde se encuentra anclada la glicoproteína E1 del virus formando un heterodímero con la glicoproteína E2 envolviendo ambas la icosaédrica de 30-35 nm de diámetro formada por unidades repetitivas de la proteína core, cuya función es proteger al genoma ARN7.
Sorprendentemente, varios estudios ponen de manifiesto la naturaleza del VHC, como una lipo-viro partícula8, con una capa gruesa de apolipoproteínas derivadas del huésped que recubren la envoltura viral, y que presumiblemente ayudarían tanto a la liberación y entrada del virus como al escape de los anticuerpos neutralizantes en la circulación sanguínea6.
Organización genómica
El genoma del VHC consiste en una única molécula de ARN monocatenario lineal de cadena simple y polaridad positiva con un único marco de lectura abierto y de aproximadamente 9.6 kilobases de tamaño, flanqueado por una región no codificante en cada extremo: hacia el 5', la región 5' NC (5' no codificante o 5' UTR) (341 bases) y hacia el 3', la región 3' NC (3' no codificante o 3' UTR) (200 bases). El genoma se encuentra asociado al core; única proteína constituyente de la cápside viral, mediante la región 5' UTR formando la nucleocápside.
La región 5' NC es conservada, con analogías superiores al 98% entre aislados y se yuxtapone con el sitio interno de entrada al ribosoma, IRES (internal ribosome entry site), regulador de la traducción de la poliproteína viral. En el extremo 5' NC, alberga dos sitios conservados que interaccionan con el miR-122 necesario para una replicación eficiente del genoma del VHC9-11posiblemente protegiendo de exonucleasas12 y de receptores de reconocimiento de la inmunidad innata, tales como RIG- I10,11,13. La región 3' NC comprende la secuencia variable de 40 bases que precede a una región rica en poli-U de longitud variable, seguida por una secuencia muy conservada de 100 bases, que también regula la traducción de la poliproteína. El marco abierto de lectura (ORF), codifica una única poliproteína precursora de alrededor 3.011 aminoácidos, la cual es traducida por ribosomas unidos al retículo endoplasmático (RE) de forma IRES-dependiente y es cortada co- y post- traduccional por peptidasas celulares y proteasas virales en al menos 10 proteínas virales maduras14.
Se ha descrito un marco de lectura alternativo (ARFP) o core+1 o proteína F (frameshift), cuya función no está clara (15). Se han detectado durante la infección por el VHC genotipo 1a anticuerpos antiproteína F, lo que indicaría que se expresa de manera natural, pero se cree que la proteína F no es necesaria para la infección y replicación16, aunque su papel en la propagación del virus y el desarrollo de enfermedad crónica no ha sido descartado17.
Las proteínas virales desde el extremo N-terminal hacia el C-terminal son las proteínas estructurales (core, E1 y E2), P7 y las proteínas no estructurales (NS2, NS3, NS4A, NS4B, NS5A y NS5B) (Figura 1).
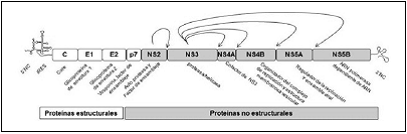
Figura 1 Organización genómica del virus de la hepatitis C. La cadena ARN positiva del genoma de 9,6-kb (parte superior), contiene un único marco de lectura abierto flanqueado por las estructuras secundarias de ARN en el extremo 5' y 3', regiones no codificante (NC). La traducción de la poliproteína precursora, mediada por el IRES, será procesa co- y post traduccional para obtener las proteínas estructurales y no estructurales (parte inferior). Se indica las funciones individuales de las proteínas del VHC.
En la porción cercana al extremo 5´ se codifican las proteínas estructurales, el core, con funciones en la formación de la cápside proteica, en la regulación, en la traducción y en la replicación del ARN y partículas de montaje; y las proteínas estructurales E1 y E2, glicoproteínas de la envoltura que participan en la adsorción y en la endocitosis mediada por receptores. La proteína integral de membrana P7 (viroporina), actúa como canal iónico dependiente de calcio en el retículo endoplásmático, y es necesaria para la morfogénesis y secreción de partículas virales infecciosas18-21.
En cuanto a las proteínas no estructurales, la proteína integral de membrana NS2, junto con NS3, constituyen la serinproteasa 2-3 que cataliza el corte entre NS2 y NS3 (con un papel importante en el ensamblaje del VHC). La proteína NS3 posee actividad de ATPasa y ARN helicasa utilizada durante la replicación y, junto a su cofactor NS4A, participa en el procesamieno de la poliproteína actuando como serin-proteasa para el corte de las proteínas no estructurales remanentes. NS4B es una proteína integral de membrana, que funciona como un andamio sobre el cual se ensamblan los componentes restantes del complejo asociado a la replicación.NS5B es la ARN polimerasa dependiente de ARN. NS5A, fosfoproteína multifuncional, es un cofactor de NS5B requerido para la replicación viral y podría estar involucrado en la resistencia al interferón22.
Una vez cortadas las proteínas no estructurales, éstas se ensamblan dentro del complejo de replicación. El VHC se replica a través de un intermediario de polaridad negativa. NS5B es capaz de sintetizar tanto ARN de cadena positiva como de cadena negativa. Las nuevas copias de ARN viral (+) interaccionan con la proteína core para formar la nucleocápside, que es finalmente rodeda por la envoltura viral que se forma por extrusión del RE. Los viriones maduros se liberan de la célula hospedadora a través de la vía secretoria celular23.
Variabilidad genética, mutación y evolución
Como otros virus ARN el VHC presenta una alta variabilidad genética y evolución rápida. Este acontecimiento se debe a que la ARN polimerasa dependiente de ARN carece de actividad reparadora de errores (ausencia de actividad exonucleasa 5'-3') provocando una acumulación de sustituciones nucleotídicas durante la replicación genómica24. Esta particularidad junto a un tamaño poblacional alto da lugar a dos niveles de variabilidad genética: intergenómica (genotipos, subtipos y aislados) e intragenómica (cuasiespecie). La tasa de error durante la replicación es de aproximadamente 10-3 a 10-5 mutaciones por nucleótido (tasa natural evolutiva de 10-4 sustituciones/ sitio/año), encontrado típicamente con la ARN polimerasa viral25, siendo la mayoría de las mutaciones generadas deletéreas26,27, combinado con la elevada tasa de producción de viriones (1010 -1012 diarios) (Neumann et al. 1998), y su corta vida media (2-5 horas) contribuyen a la variabilidad28. En un paciente infectado con VHC la población de secuencias genómicas virales que coexisten y circulan están estrechamente relacionadas entre sí (homología >98%) pero a su vez son heterogéneas genéticamente y a esa población de 29). La tasa de mutación no es homogénea a lo largo del genoma y hay regiones altamente variables (como E1 y E2) y otras regiones muy conservadas, como la región 5´NC o 3´NC30.
Genotipos y subtipos
El análisis filogenético de secuencias del VHC se ha traducido en una nomenclatura que reconoce distintos tipos y subtipos e virus. Se han definido siete grupos genéticos que difieren entre sí en más de un 30% sobre el genoma del virus completo. Estos genotipos de virus incluyen varios subtipos más estrechamente relacionados que varían en más de un 20%, mientras que dentro de cada subtipo la variación es menos del 10%31,32. El consenso del año 1994, proponía la clasificación del VHC en 6 genotipos diferentes (designados del 1 al 6) con una identidad de secuencias del orden de 69%; a su vez, cada genotipo incluía varios subtipos con una identidad cercana al 79%33. En 2005, se propone la actualización de la clasificación en genotipos y subtipos del VHC según el análisis filogenético de las secuencias de las regiones core/E1 y NS5B, y las secuencias genómicas completas4. El estudio más reciente disponible32, representa una importante actualización de la clasificación del VHC, previo consenso, con la incorporación de información adicional de secuencia derivada de más de 1.300 secuencias completas del genoma del VHC disponibles en bases de datos públicas en mayo de 2013. El análisis realizado, ha resuelto varios conflictos de nomenclatura entre las designaciones de genotipo, y el uso de criterios de consenso ha establecido la clasificación del VHC en 7 genotipos confirmados y 67 subtipos (Figura 2).
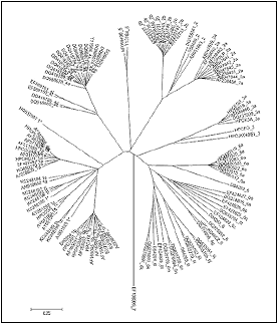
Figura 2 Árbol filogenético representativo de 118 secuencias de la región NS5B. Se construyó el árbol por neighbour-joining usando una aproximación a máxima verosimilitud, utilizando el Programa MEGA 5 (34). Las secuencias fueron elegidas (extraídas de bases de datos internacionales: GenBank (www.ncbi.nlm.nih.gov/genbank/) y Los Alamos HCV database (http://hcv.lanl.gov/components/sequence/ HCV/search/searchi.html), para ilustrar la máxima diversidad dentro de un subtipo. Los 7 genotipos etiquetados por el número de acceso y subtipo (* subtipo sin asignar). La barra de escala 0,05 representa una distancia evolutiva de 0,05 sustuciones/año.
Distribución geográfica de distintos genotipos y subtipos
Los 7 clados filogenéticos son epidemiológicamente distintos, con diferencias en el área geográfica original, en el grupo de riesgo y distribución geográfica actual, reflejando su reciente propagación epidémica a nuevos grupos de riesgo31.
Se ha relacionado a los diferentes genotipos con su distribución geográfica actual (1a, 1b, 2a, 2b y 3a distribuidos en todo el mundo y 5a, 6a y 4 sólo en ciertas regiones específicas), con la vía de transmisión de la infección (por ej. 3a y 1a predomina en drogadictos endovenosos) y la respuesta al tratamiento antiviral. En Europa y Norteamérica predomina el subtipo 1a (70%), seguido del 2b y 3a (25%)4, 35-38.
El subtipo1b tiene una distribución mundial, siendo más prevalente en Europa, China y Japón, en especial en personas de edad avanzada que contrajeron la infección mediante transfusión sanguínea antes de 199236,37. El subtipo 2a es frecuente en Japón y China35,37, el 2c en el sur de Italia39, el 4a principalmente en Egipto40, en Sudáfrica el genotipo 5 y en el Sudeste Asiático el 641,42. Se sospecha aún más, aunque con datos muy limitados que los genotipos 5 y 7 se concentran en el centro y sur de África. La alta diversidad genética que existe entre los subtipos 1a, 1b, 2a, 2b y 3a, sugiere que fueron introducidos en la población durante el siglo XX por conductas de riesgo parenterales43.
El VHC circula in vivo como distribución dinámica de genomas divergentes pero estrechamente relacionados sometidos a un proceso continuo de variación genética, competencia, y selección. definida alrededor de una secuencia "master" o consenso, representativa de las variaciones mayoritarias. La aplicación de la teoría de las "cuasiespecies" al comportamiento de los virus ARN es un tema controvertido entre los biólogos evolutivos, que se basan en el principio de la genética de poblaciones44. Aunque la población vírica que infecta a un paciente suele presentar niveles elevados de variación genética, independientemente de su hospedador30. Esta heterogeneidad genómica tiene una relevancia biológica y clínica ya que confiere una ventaja notable a la población viral permitiendo una rápida adaptación a un entorno cambiante cuando el virus está sujeto a las restricciones selectivas ejercidas por el huésped, como la inmunidad, o por factores externos, como la terapia antiviral. La gran generación de variantes o representa un desafío para el control de la infección por el VHC y tiene implicaciones importantes en el desarrollo de una vacuna y en la resistencia a antivirales, efecto que se observa durante la monoterapia con la nueva generación de inhibidores específicos de las proteínas del VHC.
Para la identificación de las "cuasiespecies" o poblaciones virales rculantes en un individuo infectado con VHC, la técnica de referencia es la clonación de los productos de ADN obtenidos mediante transcripción reversa seguida de reacción en cadena de la polimerasa (RT-PCR), y posterior secuenciación e un determinado número de clones por paciente45. Los resultados obtenidos por la técnica de clonación, nos darán una información sobre la heterogeneidad genética de las "cuasiespecies" circulantes en el paciente, descrita mediante el estudio a dos niveles: el número de variantes (número de haplotipos) presente en la población viral del paciente, y lo diferente que son entre ellos (diversidad genética). Existen diferentes opiniones en cuanto al número de clones a secuenciar. Para algunos la secuenciación de 10 variantes virales es suficiente para evaluar las virales presentes en un compartimiento46, para otros, se necesitan 20 clones para tener una representación del 95% de las variantes mayoritarias no serán detectadas47.
Con el desarrollo de las técnicas de pirosecuenciación ultraprofunda48, también es posible secuenciar una mayor proporción de las variantes víricas existentes de forma rápida, a expensas de obtener unas lecturas de menos de 400 pares de bases (pb). La ultrasecuenciación está siendo utilizada para detectar las variantes minoritarias que pudieran estar implicadas con el desarrollo de resistencias a los antivirales específicas contra el VHC y VIH49.
Algunas poblaciones minoritarias podrían ser potencialmente resistentes a drogas, y podrían estar siempre presentes cuando se inicia una terapia antiviral. Normalmente, en las "cuasiespecies" virales, se suele detectar una variante mayoritaria o "dominante" junto con las variantes que están presentes a frecuencias más bajas. La frecuencia de las distintas variantes depende de su eficacia biológica y factores del huésped50. Por lo general, la selección de muchas mutaciones tienen un coste para la eficacia biológica (fitness), aunque las variantes minoritarias pueden ser rápidamente seleccionadas durante un cambio ambiental, como la terapia con antivirales, eso no significa que perduren en la población del virus. Los factores que influyen en desarrollo de resistencias (Figura 3) incluyen: a) el número de las mutaciones necesarias para introducir el cambio de aminoácido, b) fitness del virus variante, c) la frecuencia de la variante dentro de la "cuasiespecie", d) el nivel de resistencia que confiere, e) la potencia y biodisponibilidad del agente antiviral, y f) el nivel de presión selectiva del sistema inmune, que puede provocar mutaciones de escape inmunológico que, de forma indirecta, causen resistencias51.
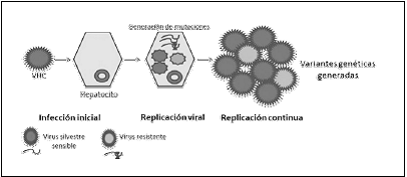
Figura 3 Generación de variantes virales de tipo salvaje y resistente, característica inherente de la infección por VHC. Después de la infección inicial de un hepatocito, el VHC comienza a replicar y con una frecuencia de alrededor 10-4 la polimerasa viral incorporará un nucleótido erróneo en el ARN, generando variantes virales. Cuando la mutación se produce en un sitio que se asocia con la resistencia a los antivirales, la variante generada puede ser resistente al antiviral.
El ciclo infectivo del VHC
El VHC infecta solamente a chimpancés y humanos, circula en suero de varias formas, viriones maduros libres, viriones unidos a inmunoglobulinas, nucleocápsides sin envoltura, y viriones fisicamente asociados a las lipoproteínas de baja densidad (LDL) o de muy baja densidad (VLDL) representando la fracción infecciosa, denominada lipoviropartícula (LVP)52. El VHC se replica principalmente en citoplasma del hepatocito siguiendo los pasos claves en su ciclo de infección: 1) Las lipoviropartículas (LVP) del VHC entran en el hepatocito a través de endocitosis mediada por receptores. 2) La proteína de la cápside y el genoma viral son liberados en el citoplasma de la célula infectada. 3) La poliproteína es procesada co- y posttraduccionalmente mediante proteasas celulares y virales para obtener 10 proteínas virales maduras. 4) La cadena positiva de ARN se replica por la ARN polimerasa dependiente de ARN a través de un intermedio de cadena negativa, las nuevas moléculas de ARN son estabilizadas mediante la unión del miR-122. 5) La cadena positiva de ARN recién sintetizada se encapsida en la nucleocápside viral (core) en estrecha proximidad a las gotas lipídicas, y las glicoproteínas de la envoltura se adquiere a través de gemación en el lumen del retículo endoplasmático. Las lipoviropartículas maduran en el retículo endoplasmático a través de interacciones con lipoproteínas, y salen de la célula a través de la ruta secretora por el aparato de Golgi y finalmente se liberan nuevos viriones53.
Vías de transmisión, prevalencia, morbilidad y mortalidad
El único reservorio conocido es el ser humano. La transmisión del VHC es fundamentalmente parenteral y puede ser vertical, en los casos de co-infección con VIH el riesgo de transmisión puede alcanzar al 6% dado que las cargas virales son más elevadas,aunque la transmisión del VHC a través del contacto sexual o de madre a hijo es ineficiente y poco frecuente54,55.
La adicción por vía intravenosa es la principal ruta actualmente de transmisión del VHC, con una prevalencia del 70- 90% en este grupo de riesgo56. La transfusión de sangre o de hemoderivados, constituye la segunda causa de transmisión, con una prevalencia del 10% en este grupo de pacientes. Actualmente el riesgo de transmisión por transfusión es de 1/2.000.000 unidades57. La transmisión dentro de la unidad de hemodiálisis parece ser el mecanismo principal de la infección nosocomial por VHC58.
Finalmente, el trasplante de órgano sólido a partir de donantes infectados es otra causa de infección por VHC, junto con las prácticas médicas no seguras59 la exposición ocupacional a sangre infectada y, posiblemente, la realización de tatuajes, piercing y acupuntura60. La transmisión por vía sexual en personas con múltiples parejas, es mayor que en parejas monógamas, siendo el riesgo elevado cuando se transmite junto con el VIH61. La seroprevalencia del VHC en HSH (hombres que tienen relaciones sexuales con hombres) oscila entre 4 y 8%, siendo mayor que la prevalencia del VHC en la población europea en general62.
Historia natural de la hepatitis C
El VHC inicia su ciclo de replicación en los hepatocitos, causando hepatitis aguda, suele pasar generalmente desapercibida n un 70-80% de las personas infectadas siendo asintomática y en un 10-20% cursa con síntomas inespecíficos (astenia, anorexia, dolor en el hipocondrio derecho) dentro de los primeros tres a seis meses de iniciada la infección. El período de incubación puede variar entre 14 a 180 días (promedio 6-7 semanas). Entre el 10-15% de los pacientes infectados resuelven favorablemente la infección, en los que el ARN-VHC en suero se vuelve indetectable y los niveles de Alanina transaminasa (ALT) retornan a la normalidad, pero la mayoría de los individuos VHC positivos (> 80%) desarrollan una infección persistente a pesar de la presencia de anticuerpos séricos contra el mismo y de una respuesta de las células T citotóxica antiviral multi-específica, debido tanto a la alta tasa de mutación, a su estructura genómica en cuasiespecie y a su elevada producción de viriones (1012 viriones/día, con una vida media del virión de sólo 2,7 h). La mayoría de estos individuos evolucionan hacia la cronicidad, padecerán de fatiga y presentarán niveles séricos de ALT elevados o fluctuantes, mientras que un tercio presentará niveles persistentes normales, a pesar de la viremia y la lesión hepática continua. Una vez establecida la infección crónica, de ellos un 10% desarrolla insuficiencia hepática, 20% muestra signos de cirrosis en un período de 15 años desde su infección, el 13-15% desarrolla una descompensación hepática y entre el 2-7% sufre un hepatocarcinoma celular63. La mortalidad global oscila del 4-9% y el estadío terminal de la infección crónica por VHC es una de las principales indicaciones de trasplante hepático64. Desafortunadamente, la infección crónica recurrente se establece de manera prematura en el órgano trasplantado, y se acelera la progresión de la enfermedad65.
Tras la exposición al virus, el ARN-VHC puede ser detectado en suero entre la y 2a semana. Los niveles séricos de ALT (indicador indirecto de inflamación hepática) aumentan hasta 10 veces superior a lo normal 2-8 semanas después de la exposición al virus, debido a la lisis de los hepatocitos infectados mayoritariamente por los linfocitos T CD8+ específicos, generalmente con un patrón fluctuante (Figura 4).
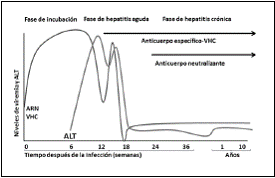
Entre los pacientes con infección crónica, el riesgo de cirrosis tras 20 años de infección varía entre 10-15% para los hombres y 1-5% para las mujeres, la razón de esta diferencia no se conoce, algunos estudios muestran estimaciones de hasta l 50%66. Una vez que se establece la cirrosis, la tasa de desarrollo de carcinoma hepatocelular es de 1-4% por año67.
Tras la infección aguda, las células T específicas del VHC son normalmente detectables 5-9 semanas después de la infección68, y los anticuerpos específicos del VHC son detectados 8-20 semanas después de la infección 69. El título de anticuerpos se reduce en 10-20 años, aunque la respuesta específica de las células T CD4+ y CD8+ persiste incluso después de varias décadas 70.
La infección por el VHC induce grandes cambios en el metabolismo e los lípidos celulares, incluyendo una reducción de los niveles de lipoproteínas séricas y una acumulación de lípidos en las células parenquimatosas del hígado (esteatosis) (VHC genotipo 3a)71,72.
Diagnóstico
La presencia de anticuerpos contra el VHC indica que una persona ha sido infectada. El diagnóstico de la infección crónica se realiza cuando los anticuerpos para el VHC están presentes en la sangre durante más de seis meses. La infección aguda a menudo no es sintomática, y los anticuerpos IgG anti- VHC suelen ser negativos durante la fase aguda y la infección sólo se puede determinar detectando en suero el ARN del VHC73. Otras pruebas útiles para integrar la presunción diagnóstica, son las pruebas de funcionalidad hepática, los niveles elevados de las enzimas ALT y AST (aminotransferasa de aspartato), y la FA (fosfatasa alcalina) y la GGT (gamma glutamil transferasa), indicadoras de daño hepático y alteraciones del tracto biliar, así como otras anomalías74.
Diagnóstico indirecto
El ELISA, método de diagnóstico indirecto serológico basado en el estudio de la respuesta inmune específica frente al VHC mediante la detección de anticuerpos circulantes, que se utiliza como cribado y diagnóstico de primera línea. Indica exposición al virus sin diferenciar infección aguda, crónica o resuelta.
Las pruebas confirmatorias de los resultados positivos del ELISA basadas en la técnica de "Western blot", también conocido Recombinant inmunoblot assay (RIBA-3) de tercera generación, como han quedado obsoletas debido a la mejora de la sensibilidad de los sistemas de ELISA de tercera generación y a la aparición de técnicas de detección del ARN-VHC circulante que tienen una elevada sensibilidad y especificidad75,76.
Diagnóstico directo
Se basa en la detección de componentes virales, como diferentes fragmentos del genoma del VHC (ARN-VHC) o determinadas proteínas virales, esto implica la existencia de replicación vírica activa. Las técnicas de diagnóstico molecular permiten una detección tanto cualitativa como cuantitativa del ARN-VHC circulante en suero, plasma o tejido.
Diagnóstico cualitativo (ARN-VHC)
El ARN-VHC es detectable entre la primera y segunda semana después de la infección, y se utiliza para predecir precozmente la respuesta virológica sostenida (RVS), también para el diagnóstico durante el periodo de ventana serológico, para detectar la infección en pacientes inmunocomprometidos que no hayan desarrollado la repuesta inmune, y para el diagnóstico precoz en la infección perinatal.
Diagnóstico cuantitativo (viremia)
Las técnicas cuantitativas permiten cuantificar la carga vírica basal y evaluar la presencia de respuesta virológica temprana (RVT) a las 12 semanas del tratamiento. La RT-PCR en tiempo real es la técnica de elección para detectar y cuantificar el ARN-VHC en la práctica clínica75-77.
La detección cuantitativa del antígeno core del VHC con la técnica ELISA, se utiliza para el diagnóstico precoz de la infección aguda, pero la desventaja de este método es que tiene una sensibilidad menor al 95% (niveles de ARN-VHC inferiores a 20.000UI/ml no detecta), y una especificidad cercana al 100% que las técnicas de diagnóstico molecular, pero su coste-eficacia, facilidad de utilización y ausencia de contaminantes hacen que sea una alternativa para la detección de viremia en zonas geográficas donde no disponen de la tecnología molecular78.
Genotipado del VHC
El genotipo de VHC que ha infectado al paciente, es un factor importante para tener en cuenta sobre el pronóstico y duración del tratamiento (generalmente puede ser más corto para los pacientes infectados con los genotipos 2 o 3 en comparación con los pacientes infectados con los genotipos 1 o 479, el fármaco, la dosis y la probabilidad de responder al tratamiento antiviral80. En cuanto al significado clínico a nivel de subtipo está teniendo importancia con la terapia que actualmente se trata de establecer, en aquellos pacientes que no responden al tratamiento estándar, el interferón- (peg-IFN- ); y en aquellos pacientes naive genotipo 1, con los nuevos antivirales de acción directa (AAD)80. Finalmente el genotipo del VHC es relevante para el tratamiento con peg-IFN- con AAD53.
La determinación del genotipo del VHC, basada en la secuenciación de la región no codificante 5' NC o de la región NS5B, mediante técnicas in house seguido de un análisis filogenético de las secuencias obtenidas junto a las secuencias prototípicas, es considerada la técnica de referencia, pero su complejidad técnica hace que solo en centros con personal cualificado puedan utilizarla81.
Aunque la región 5' NC es altamente conservada entre los genotipos, pero con diferencias en la secuencia de nucleótidos específicas para cada genotipo y los principales subtipos, la discriminación de ciertos genotipos (aislados del genotipo 6 han sido identificados como genotipo 1) y entre subtipos (1a versus 1b y 2a versus 2c) no siempre es fiable al utilizar esta región. Esto se debe a la elevada homología que presentan entre sus secuencias; los subtipos 1a y 1b difieren en un nt (A/G) en la posición 99 y los subtipos 2a y 2b en dos nt en la posición 124 y 164 respectivamente. Sin embargo, la secuencia de la región core o NS5B es más fiable ya que tienen una variabilidad genética suficiente para poder diferenciar entre subtipos77,82.
Epidemiología de la infección por VHC
Se estima que 200 millones de pacientes en todo el mundo están infectados con el VHC y que aproximadamente 130- 150 millones (3% de la población mundial) vive con hepatitis crónica. Alrededor de 3-4 millones de personas son infectadas por año, y más de 350.000 personas mueren cada año a partir de enfermedades relacionadas con la hepatitis C (81). La seroprevalencia de la infección del VHC varía geográficamente (Figura 5). El VHC tiene una incidencia de 8,7 por 100.000 en los Estados miembros de la Unión Europea (UE) con alta prevalencia en usuarios de drogas inyectables83. Se estima que hay 2-5 millones de personas infectadas con VHC en Europa. La incidencia ha tendido a aumentar durante los últimos años, además la inmigración procedente de África Subsahariana, Asia y Pakistán, donde la prevalencia es muy elevada (9-11%) ha contribuido al incremento de la prevalencia en Europa36,84. La alta prevalencia se debe también a la propagación iatrogénica en la población de edad avanzada que tuvo lugar hace más de medio siglo, seguido 3 décadas más tarde por otro aumento de la prevalencia en individuos jóvenes por la adicción a drogas por vía parenteral.
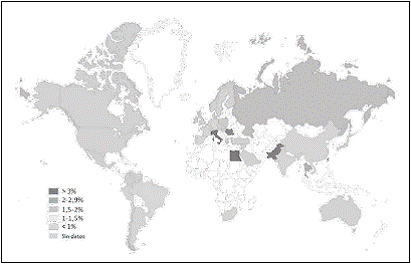
Las tasas han disminuido en el mundo occidental desde la década de 1990 debido a la detección del VHC en sangre antes de la transfusión85. En los EE.UU. se estima un aumento significativo del 24% en la prevalencia de cirrosis y carcinoma hepatocelular de los casos reportados en ese país (86). El VHC causa el 27% de las cirrosis y el 25% de los casos de carcinoma hepatocelular en el mundo, y principal causa de trasplante hepático de EE.UU. y Europa occidental87. En Europa la alta prevalencia de la infección de VHC se registra con un 53 % en el grupo de 25 y 44 años de edad, y el 64,4% son hombres88.
Conclusiones y Perspectivas
La infección crónica provocada por el VHC sigue siendo un gran problema de salud pública a nivel mundial. Se estima que 200 millones de personas (3% de la población mundial) se encuentran infectadas por este virus. La hepatitis crónica C supone la primera causa de muerte de origen hepático y la primera indicación para el trasplante hepático en los países desarrollados.
El VHC tiene una alta variabilidad genética, asociada a diferencias en la progresión de la enfermedad, y en la eficacia del tratamiento antiviral. Hoy en día esta enfermedad cuenta con más afectados que el VIH, sin embargo como la inmensa mayoría de los casos no están diagnosticados, se considera que la cifra es mucho mayor y de ahí que se le llame Es por ello que el sistema de salud pública precisa de medidas enfocadas a un diagnóstico precoz y prevención de la transmisión.
El tratamiento basado en la combinación de dos fármacos no específicos peg-IFN-, provoca numerosos efectos secundarios, el tratamiento es de larga duración y además es costoso y tiene poco éxito en pacientes infectados con genotipo 1. Por otra parte, la reinfección del injerto post-trasplante es universal, ocasionando una hepatitis más agresiva y más difícil de tratar con peg-IFN- Esto ha impulsado al desarrollo de nuevos fármacos más específicos, los AAD que están dirigidos contra proteínas del virus, como la proteasa NS3, la proteína NS5A o la polimerasa NS5B, y han mostrado una gran efectividad con mínimos efectos secundarios. A pesar de su alto precio, en pacientes con enfermedad avanzada o trasplantados el acceso a los nuevos tratamientos con ADD es fundamental para mejorar su pronóstico y detener la progresión de la enfermedad.

