











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
La fiebre aftosa, es una epizootia que la causa un Aphtovirus de la familia Picornaviridae1,2,3. La cual se caracteriza por fiebre y formación de aftas principalmente en la cavidad bucal, hocico, espacios interdigitales y en las pezuñas, patología característica en los ungulados4.
El ARN de este virus contiene un único marco de lectura abierto que codifica para una larga poliproteína que es posteriormente clivada durante la traducción. Estos clivajes son llevados a cabo mayoritariamente por proteasas codificadas por el virus (Lpro, 2C, 3C). El genoma del virus de la fiebre aftosa (VFA) se divide en cuatro regiones codificantes denominadas L, P1, P2 y P3. La región L, ubicada en el extremo 5 ́, codifica para un polipéptido líder que se autocliva del extremo amino terminal del precursor de las proteínas de la cápside entre una lisina y una glicina. Hacia el extremo 3 ́ de la región L se halla la región P1, que codifica para las cuatro proteínas estructurales en el orden VP4, VP2, VP3 y VP15.
Con respecto a las regiones P2 y P3, que codifican a las proteínas no capsidales (PNC) del virus, se conoce que intervienen dentro del proceso de replicación6. En la región P2 se encuentra la proteína 2B, que es determinante dentro del rango del hospedador y la proteína 2C, que se encuentra involucrada en la síntesis de ARN con posible actividad helicasa y se encuentra altamente conservada7. En la región P3 se encuentra una proteasa 3C, responsable de la mayoría de los clivajes del procesamiento primario y secundario de la poliproteína precursora8.
En esta región se encuentra una ARN polimerasa viral llamada 3D, que es ARN dependiente y es conocida como antígeno VIAA (del inglés viral infection associated antigen, antígeno asociado a la infección viral), posee una secuencia de nucleótidos y aminoácidos (a.a), altamente conservada en todos los serotipos y subtipos del VFA9,10.
Las proteínas no capsidales se utilizan como marcadoras cuando existe sospecha de la presencia del virus, y únicamente se determina indirectamente midiendo presencia de anticuerpos contra las PNC, por medio de la prueba screening I-ELISA 3ABC y se confirma por medio del ensayo inmunoenzimático de electrotransferencia (EITB), pruebas desarrolladas por el Centro Panamericano de Fiebre Aftosa (PANAFTOSA) (11,12.
Actualmente, la Organización Mundial de Sanidad Animal (OIE) suspendió el estatus del país como libre de aftosa con vacunación, por algunos focos que se han venido presentando de la enfermedad. Los entes de control utilizan estrategias para controlar la enfermedad y como herramienta para discriminar animales vacunados de enfermos, utilizan esta prueba indirecta para demostrar ausencia de inmunogenicidad contra las PNC13, por lo cual los laboratorios productores de la vacuna deben descartar estas proteínas de sus productos finales. Esta normatividad exigida por la Organización Mundial de Sanidad Animal (OIE) y por PANAFTOSA, debe ser acatada por cada ente de control de los países miembros12,13,14,15.
En Colombia el Instituto Colombiano Agropecuario (ICA), se encarga de que se cumplan todos los parámetros establecidos15. Los lineamientos que se deben seguir cuando un lote de vacunas es fabricado, constan básicamente de evaluar estas suspensiones en bovinos que se encuentren en una zona libre de aftosa sin vacunación y que con anterioridad se les haya realizado la prueba para determinar la ausencia de IgG contra las PNC, lo cual indica que no han tenido contacto con el virus. Técnicamente el procedimiento comienza inmunizando con la vacuna 30 bovinos, los cuales son revacunados 30 días después; el suero se analiza el día 60, con el mismo kit I-ELISA 3ABC, para determinar la ausencia de IgG contra las PNC, si hay presencia de anticuerpos los lotes son rechazados12,13,14. Sin embargo, por los enormes costos económicos es poco probable que se pueda llegar a un proceso de purificación que pueda eliminar el 100% de las PNC, ya que se elevaría el precio de la vacuna considerablemente, haciéndola inalcanzable para la mayoría de los ganaderos.
No obstante es importante que las empresas puedan monitorear por medio de un kit directo la cantidad de remanente de las PNC, de tal manera que las puedan llevar a un nivel menor al 5% en los lotes de vacunas, minimizando una reacción inmunológica, además es una estrategia que serviría como prueba confirmatoria, dejando a la prueba indirecta como complementaria.
El objetivo de esta investigación, fue purificar y caracterizar la PNC 3D, ya que, su alta pureza, facilitará la producción de IgG específicos con miras a producir una prueba que cuantifique la PNC polimerasa 3D, en las vacunas existentes en el mercado nacional e internacional.
Materiales y métodos
Obtención del antígeno PNC 3D El antígeno fue aislado de una suspensión de virus inactivado facilitado por el Laboratorio LIMOR DE COLOMBIA, la cual consistió en una suspensión bivalente del VFA elaborada con los serotipos A24 Cruzeiro y O1 Campus cultivados en células BHK-21 (Riñón de Hámster Lactante).
Proceso de purificación de la PNC 3D
Diálisis y cromatografia de Intercambio catiónico. El cultivo de virus, se concentró en una celda de ultrafiltración AMICON®, con una membrana Millipore® de 10.000 Daltons (Da), La muestra de 1.200 ml se llevó a 60 ml con una concentración final de 32 mg / ml. En la misma celda se dializó la muestra 4X con 240 ml de solución tampón (20 mM de Na2HPO4 y 20 mM de NaCl con Tween) pH 7.4, se alícuoto y se guardó a -20ºC.
De la solución dializada se inyecto 160 mg (5 ml) a la columna de Sepharosa-DEAE (2.5 × 10 cm) previamente equilibrada con la misma solución utilizada en la diálisis y que a su vez fue utilizada para lavar la columna después de inyectada la muestra, hasta que la Absorbacia a 280 nm (A280) cayó a 0.0. Las proteínas retenidas en la columna fueron eluidas con un gradiente salino de 20 mM a 800 mM de NaCL en la misma solución tampón y colectado en fracciones de 7.5 ml por tubo, la concentración de cada tubo fue cuantificada a A280, en un equipo NanoDrop 2000. Las fracciones se unieron por picos, se tomó la A280 y se determinó la concentración final, adicionalmente, para corroborar los resultados, se realizó el método de ácido acido bicinconinico16
Electroforesis SDS-PAGE. La electroforesis SDS-PAGE al 15% se realizó de acuerdo al método de Laemmli17. Cada uno de los tres picos se mezclaron con el tampón de muestra (20% Glicerol, 2% SDS, 0.5 M Tris-HCl pH 6.8, 10% β-mercaptoetanol, y 0.05% de azul de bromofenol) en un volumen 1:1 (vol / vol); luego fueron tratados térmicamente por 5 min a 100 ºC. De cada pico se aplicó 10 μg en los respectivos pozos del gel y se corrió a 200 Voltios. Las proteínas fueron visualizadas utilizando la coloración azul brillante de Coomassie.
Los pesos moleculares de las bandas se calcularon utilizando el software Image LabTM, utilizando como referencia un marcador de peso molecular de bajo rango de Biorad: Fosforilasa b (97.4 KDa), seroalbúmina bovina (66.2 KDa), ovoalbúmina (45 KDa), anhidrasa carbónica (31 KDa), inhibidor de tripsina de soja (21.5 KDa) y lisozima (14,4 KDa).
Electroforesis bidimensional. Para realizar la electroforesis en dos dimensiones (2D) (18, la muestra de PNC 3D (50 mg), se disolvió en un tampón de rehidratación (Urea 8M, CHAPS 2 %, DTT 50 mM, Anfolitos 3/10, trazas de azul de bromofenol en agua desionizada), hasta completar 125 μl, volumen recomendado de carga para una tira IPG de 7 cm, que contenía un rango lineal de pH de 3 a 10; esta hidratación se llevo a cabo por 12 horas.
Después de la rehidratación, se llevó a cabo el isoelectroenfoque, usando el equipo PROTEAN 12 IEF SYSTEM de Biorad, utilizando el siguiente protocolo: 20 min a 250 V, 2.5 h a 4000 V, hasta alcanzar 10.000 V / hora. Las tiras IPG (Immobilized pH gradient), se sometieron a reducción y alquilación, incubándolas durante 10 min en una solución con 8 M de urea, 30% de glicerol, 2% de SDS y 125 mM de DTT, y 10 min en la misma solución tampón en la que se reemplazará el DTT por 125 mM de iodoacetamida. Después de esto se realizó la separación de proteínas de acuerdo con su peso molecular colocando la tira equilibrada sobre el gel de poliacrilamida al 12.5 % en presencia de SDS y cubierto con overlay agarose. La electroforesis se llevó a cabo usando el sistema Miniprotean a un voltaje de 200 Voltios. Se tiñó con nitrato de plata y se documentó con el sistema Gel Doc XR+ de Biorad.
Secuenciación de las proteínas. El procedimiento se realizó por espectrometría de masas MALDI-TOF y Electroespray trampa iónica (LTQ_Velos) y se llevó a cabo por el servicio de proteómica del Centro de Biología Molecular Severo Ochoa (CBMSO), en Madrid España (19, 20, 21).
Inmunización en cabras con la proteína no estructural 3D. Este estudio se realizó con el aval del Comité de Ética de la Universidad de Pamplona del 25 de mayo de 2015, según acta 003 y con la colaboración de un médico veterinario en la finca Villa Marina de propiedad de la Universidad de Pamplona, se diseñó un esquema de inmunización para dos cabros con la proteína purificada 3D. La cantidad del inmunógeno utilizado fue de 200 mg / ml. Cada animal fue inmunizado con 2 ml (400 mg), antes de la primera inmunización se le tomaron pruebas de sangre a los cabros. La mezcla del inmunógeno con el adyudante incompleto de Freund fue administrado por vía subcutánea los días 0, 7, 14 y 21, después de 15 días desde la última inmunización 22, se le extrajo al animal una bolsa de 400 ml de sangre, con anticoagulante. El suero se obtuvo por centrifugación de la sangre a 700 G gravedades / 10 minutos a +4ºC, en una centrífuga KR25i Jouan de mediana velocidad, el suero fue decantado y conservado a -20ºC hasta su utilización.
Purificación de anticuerpos policlonales. El plasma del animal inmunizado, se precipitó con sulfato de amonio en polvo, aplicándole 27 gramos por cada 100 ml de muestra, a una temperatura de 3-4ºC, el cual fue agregado en pequeñas cantidades y en constante agitación. Luego se centrifugó a 6.000 g por 15 minutos, se descartó el sobrenadante23,24,25. El precipitado se lavó tres veces con una solución de sulfato de amonio 2 Mol/L y se centrifugó bajos las mismas condiciones que antes. El precipitado se disolvió en 200 ml de agua, y se transfirió la solución a una membrana para diálisis, (Dialysis tubing de Fisherbrand™). La muestra se dializó alternadamente así, primero por 12 horas con agua y luego por 12 horas con 70 mM de NaAc pH 5.5, dos veces cada uno, luego se centrifugó. Al sobrenadante se le ajustó el pH a 8.5 con 0.4 M NaHCO 3, 2.0 M Na2CO3, se tomó la A280, y la concentración se determinó utilizando como coeficiente de absorción 1.3826.
La solución proteíca fue inyectada a una columna que contenía 2 ml de Sefarosa-proteina A (0.8 x 4 cm). La columna se lavó con la solución tampón (10 mM sodio fosfato, 0.15 M NaCl, pH 8.2). hasta que la A280 fue 0.0 y las IgG fueron eluidas de la columna con la solución tampón 0.1 M sodio citrato, pH 3.0. La solución de las IgG fue neutralizada con 1 M de NaHCO3, a un pH entre 6 - 727.
La discriminación de las IgG policlonales contra la proteína 3D se hizo en una columna de propileno de 1.5 x 12 cm, con la matriz de afinidad de Sefarosa-proteina-3D, producida por el laboratorio, con Sepharosa activada CNBr 4B, de acuerdo con las instrucciones del fabricante. La matriz de Sefarosa-proteina- 3D, se equilibró con 20 mM fosfato y 100 mM NaCl pH 7.2; se le adicionaron 10 ml de la solución de IgG (14 mg) y se lavó con la misma solución tampón hasta que A280 fue de 0.0, la elución se hace con 100 mM de glicina / HCl pH 2.7, la solución de las IgG fue neutralizada con 1 M de NaHCO3, a un pH de 6.0.
Inmunodifusión. La especificidad de las IgG policlonales, se evaluó mediante la técnica de inmunodifusión doble 28. Brevemente, se utilizaron placas con agar al 1%, a las que se les realizaron una serie de perforaciones distribuidas en forma equidistante. Las perforaciones se llenaron en el centro con 10 ml del antígeno, proteína 3D purificada (10 mg) y en las cuatro esquinas con 10 ml del IgG (10 mg). Las placas se incubaron a 4 º C, por un tiempo estimado de 24 a 48 horas en cámara húmeda.
Western Blot. Después de realizar la electroforesis de la proteína 3D en las condiciones previamente descritas, se hizo la transferencia de esta, a una membrana de PVDF (polifluoruro de vinilideno), usando una cámara Mini Trans-Blot® Cell con solución tampón de transferencia (25 mM Tris-HCl pH 8.3, 190 mM glicina, 20% metanol, 0.1% SDS) a 100 voltios por 8 horas a 4ºC29.
Posteriormente la membrana se bloqueó con una solución tampón que contenía 20 mM de fosfato, 150 mM de cloruro de sodio y albumina al 3 % BSA (PBS-BSA) por una hora a temperatura ambiente. Luego de lo cual la membrana fue incubada con la IgG contra la proteina 3D diluida (1:1000 en PBS-BSA) a una concentración final de 1 mg / ml, y fue incubada durante 3 horas a 37 °C. Luego se adicionó la anti IgG conjugada con peroxidasa del rábano, HRP (por sus siglas en inglés) de Sigma aldrich, diluida 1:3000 con PBS-Tween 20, se incubó por 1 hora bajo constante agitación. Se reveló con un kit comercial de substrato de peroxidasa (OPTI-4CN 4- Cloro naftol) de Biorad. La reacción se detuvo agregando agua desionizada.
Resultados
El pelfil cromatográfico obtenido para el proceso de purificación de la PNC 3D, mostró tres picos: El pico I, se ve divido en dos, pero ambos mostraron el mismo contenido proteico en la electroforesis por lo que se estableció como uno solo, el cual corresponde a las proteínas que no interaccionan con la matriz de sefarosa-DEAE, eluyendo directamente con la solución tampón de lavado; los picos II y III corresponden a las proteínas retenidas en la matriz, las cuales eluyeron con el gradiente salino que va de 20 a 800 mM de Cloruro de sodio (Fig 1A).
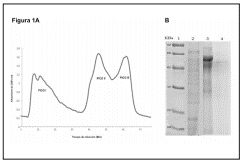
Figura 1 A. Perfil cromatográfico obtenido de la purificacion de la proteína PNC 3D a partir del cultivo de virus del serotipo VO1C del virus de la fiebre aftosa, Pico 1 (Proteinas no retenidas en la matriz); Pico 2 y 3 (Proteinas eluidas por el gradiente salino). B. Electroforesis SDS PAGE al 12.5%. Carril 1: Marcador de peso molecular (kDa) ; Carril 2: Proteínas obtenidas en el Pico I; Carril 3: Proteínas obtenidas en el Pico II ; Carril 4: Proteínas obtenidas en el Pico III
En el pico I de la figura 1A, se observan 10 bandas con pesos moleculares heterogéneos entre 20 a 70 kDa, con una cantidad de 8 mg (Carril 2, Figura 1B), todas las bandas presentes fueron identificadas por la tecnica de MALDI-TOF, como pertenecientes al hospedador del orden rodentia. El pico II, se le determinó una masa proteica de 10 mg de proteina total, las cinco bandas presentes fueron identificadas como albumina bovina (Carril 3 Figura 1B), inclusive las de menor peso molecular, clara indicación de degradaciones enzimáticas en la solución.
A el pico III, se le determinó una masa proteica de 4.8 mg; en la electroforesis se observa una sola banda (Carril 4, Figura 1B); para demostrar aún mas su pureza, este pico fue teñido con nitrato de plata que es mucho mas sensible, demostrándose igualmente la misma banda sin contaminantes ( Carril 2, Figura 2A), su identificacion no se logro por MALDI-TOF pero si fue identificada por la técnica de Electroespray trampa iónica (LTQ_Velos), como RNA-directed RNA polymerase 3D-POL (P3D-POL), con la secuencia SLGQTITPADK (Figura 2B), ubicada entre los aminoácidos 2222 al 2232 que se encuentra dentro de la longitud de los 470 a.a que abarcan a esta proteína. Además su peso molecular fue determinado en 51 KDa, utilizando como referencia un marcador de peso molecular conocido y el software Image Lab™ (Figura 3). Este peso tiene gran similitud con el de otros trabajos donde esta proteína es producida de forma recombinante con un peso molecular de 53 KDa30.
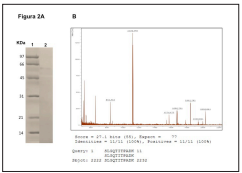
Figura 2 A. SDS PAGE de la proteína no capsidal 3D teñida con nitrato de plata. B. Espectro de masas que coincide con la masa teórica de la proteína no capsidal 3D polimerasa.
La electroforesis bidimensional de la proteína 3D, ya purificada, se realizo usando isoelectroenfoque (IEF) en la primera dimensión y un gel denaturante (SDS-PAGE) en la segunda dimensión (Figura 4), revelando características interesantes de la proteína, donde se observa claramente tres isoformas, en un rango de pH aproximado entre 5 y 6, abarcando el área del punto Isoeléctrico teórico de la proteina 3D (pH 5.96), el cual concuerda con el suministrado por la base de datos Uniprot.

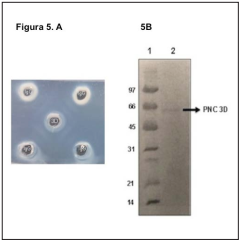
La discriminación de los anticuerpos específicos contra la proteina 3D purificada, se logro por medio de una cromatografia de afinidad (Figura 5, carril 2 y 3); donde no se observan impurezas ni en 5 ni en 10 mg, se obseva solo la cadena pesada y liviana que las caracteriza con pesos moleculares de 50 y 25 kDa.
Figura 5 SDS-PAGE de los anticuerpos purificados por cromatografía de Afinidad. Carril1: Marcador de peso molecular. Carril 2 y 3: IgG cargada con 10 mg y 5 mg
El test para detección de IgG específicos (método de Outcherlony) mostró dos bandas de precipitación tipo romboidal, para las IgG del suero hiperinmune de las cabras obtenido en la inmunización, lo que indica que hubo interacción entre la proteina-3D y su anticuerpo específico, (figura 6A). El cual fue confirmado por el revelado de la banda que hizo reacción con la técnica de western blot (Figura 6B)
Discusión
La elección de la proteína no capsidal 3D polimerasa para su purificación, se debió a que además de estar directamente implicada en la replicación del virus y de ser una proteína conservada entre los diferentes serotipos, también es altamente inmunogenica3, lo que fue importante para la obtención de los anticuerpos específicos contra esta proteína. Su purificación de forma nativa la hace un método óptimo por la abundante materia prima del cual se puede obtener la proteína, lo que baja los costos comparándolo con la purificación de forma recombinante, pues además de este modo se han encontrado problemas asociados, como la presencia de anticuerpos contra los antígenos utilizados como vectores de expresión, lo cual puede resultar en significativas reacciones no especificas30.
En esta investigación se logró de manera exitosa el aislamiento y la purificación de la proteína de interés utilizando el método cromatográfíco de intercambio iónico, quedando está retenida en la matriz y siendo específicamente eluida en el pico III, gracias a él gradiente salino utilizado; siendo aislada de las impurezas que quedaron en los otros dos picos. Durante todo el tiempo de la investigación se observó que esta purificación depende mucho del estadio de maduración del virus; pudiéndose demostrar que solo cuando el cultivo de virus se encuentra en un estadio maduro, es donde se logra purificar, muy posiblemente porque ya la poliproteína está totalmente clivada y las proteínas individualizadas listas para cumplir sus funciones biológicas.
La pureza tanto del antígeno no estructural 3D como de los anticuerpos específicos contra ella, abre la posibilidad de producir un kit de Elisa directo, que sirva para evaluar las suspensiones en las distintas etapas del proceso de elaboración de la vacuna anti-aftosa, con el objetivo de que los productores mejoren sus procedimientos y logren reducir por debajo del 5% la presencia de PNC en la vacuna; todo esto con el fin de seguir los lineamientos establecidos por los entes de control internacionales de la enfermedad y tener una vacuna de alta calidad, que no genere interferencias en los métodos de diagnóstico de la patología y se logre diferenciar los animales vacunados de los infectados.

