











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkEn los últimos años Colombia ha experimentado cambios significativos en sus frecuencias y en las cargas delas enfermedades, haciendo que la patología genética se convierta en una de las principales causas de morbimortalidad, sobre todo infantil 1,2. Según datos del DANE la primera causa de mortalidad infantil en el país desde 1977 es el grupo denominado "afecciones originadas en el periodo perinatal" con el 43 % de los fallecimientos. La segunda causa, desde 1994, la constituyen las anomalías congénitas con el 11 % de los niños fallecidos, indicando que el 54 % de los decesos pediátricos antes del primer año de vida puede ser explicado por causas genéticas 3,4. Se estima que en alrededor del 40 % de los casos de discapacidad, en cualquiera de sus grados, una etiología de origen genético es detectable; esto soporta el impacto de las alteraciones genéticas dentro de la morbimortalidad no solo infantil sino también a largo plazo 5.
La gran mayoría de las patologías genéticas hacen parte del grupo de enfermedades huérfanas, un grupo de patologías que cuentan con reconocimiento especial dentro del marco legal Colombiano 6. Sin embargo, similar a lo descrito en otros países en vías de desarrollo, la mayoría de la población colombiana tiene un acceso muy limitado a los servicios de Genética Médica por factores económicos, geográficos, de distribución de la prestación del servicio, así como la reducida disponibilidad de personal de la salud con entrenamiento especializado, retrasando una oportuna atención a este grupo de pacientes y familias 7,8.
Existen reportes entre la comunidad médica nacional sobre la presencia de zonas geográficas con mayor frecuencia de enfermedades de origen genético ya sea por antecedentes de efectos multifundadores, como es el caso de Antioquia ( ), o por posible endogamia y consanguinidad como se ha visto en el altiplano Cundi-Boyacense. Esta apreciación es soportada por los resultados de Gómez y Wilches 9,10, quienes lograron definir frecuencias mayores a las esperadas para algunas enfermedades recesivas como Mucopolisacaridosis y detectaron un posible haplotipo ancestral común en Enfermedad de Gaucher.
La gran afluencia de pacientes provenientes de Boyacá a la consulta clínica de la Maestría de Genética Humana de la Universidad Nacional junto a la limitada información sobre la carga de la enfermedad genética en este departamento, generó la necesidad de realizar una exploración inicial de la patología genética empleando una metodología de medicina comunitaria que en principio pudiese eliminar algunas barreras de acceso y ampliar el conocimiento epidemiológico de esta región del país.
MÉTODOS
Con base en la información referida por algunos neuropediatras y pediatras de Boyacá, los datos referentes a la densidad poblacional en el Departamento y por información extraída de las historias clínicas de pacientes de la consulta clínica de la Maestría de Genética Humana de la Universidad Nacional, se seleccionaron algunas regiones de Boyacá para la realización de trabajo de campo. Se visitaron las cabeceras municipales de Sogamoso, Soatá, Chiquinquirá, Garagoa, Cocuy y Tunja, convocando a la población de estudio en las instituciones prestadoras del servicio de salud (cobertura total de aproximadamente 400 000 habitantes). La actividad consistió en dos visitas: la primera realizada en año 2013 y la segunda en año 2014.
En cada visita se llevó a cabo, primero una sesión de sensibilización sobre las enfermedades genéticas al personal de salud y segundo, la ejecución de jornadas clínicas en búsqueda de pacientes con sospecha de alguna patología genética, donde se levantaron datos epidemiológicos, demográficos y clínicos con la ayuda de un grupo de médicos especialistas (genetistas, neuropediatra y genetista bioquímico) y un grupo de maestrantes de Genética Humana de la Universidad Nacional.
Los diagnósticos encontrados en la jornada se clasificaron en 5 grupos:
- Cromosómico, que incluye rearreglos, deleciones, duplicaciones y anomalías numéricas.
- Monogénico, para desordenes conocidos con un gen causante.
- Multifactorial, condiciones para las cuales interacciones gen-gen, gen-ambiente, han sido descritas o sugeridas en la literatura previamente.
- Ambiental, pacientes con secuelas de noxas prenatales externas, sin compromiso genético.
- Idiopático. En este grupo clasificamos condiciones con alta sospecha de etiología genética, en la cual no se dispone de una prueba molecular específica para su diagnóstico.
Además se realizó otra caracterización:
Pacientes en Estudio: es decir, aquellos con manifestaciones inespecíficas pero sugestivas de alguna entidad genética sindrómica y que no han sido estudiados previamente, siendo difícil su diagnóstico por sólo anamnesis y examen físico. A estos pacientes se solicitaron estudios de extensión según el caso.
El análisis estadístico se realizó con una descripción de las principales variables socio-demográfica y clínica de los pacientes.
RESULTADOS
Se evaluaron 152 pacientes que habían sido referidos por los médicos tratantes (Tabla 1). Empleando anamnesis y exámenes clínicos detallados se realizaron los respectivos diagnósticos clínicos. En aquellos pacientes que habían sido diagnosticados previamente, se reevaluó la información disponible y se brindó la asesoría genética, recomendando seguimiento a cada caso.
Tabla 1 Número total de pacientes valorados en las dos jornadas (2013-2014) de exploración de patología genética en seis centros de Boyacá
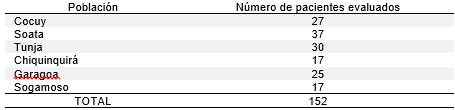
La media de la edad de los pacientes fue de 14,6 años con un rango desde los 0,1 años (1 mes de edad), hasta los 70 años de edad. De los 152 pacientes, seis (3,9 %) contaban con pruebas diagnósticas confirmatorias de su patología genética (Tabla 2).
Tabla 2 Clasificación de los diagnósticos encontrados en 152 pacientes de los 6 centros de referencia del Departamento de Boyacá

(SOSP) Sospecha de la enfermedad;*diagnóstico de enfermedad genética confirmado por prueba específica;**diagnóstico confirmado en uno de esos casos por prueba enzimática; ***sospecha de EIM;[AD] Autosómico dominante; [AR] Autosómico recesivo; [XL] Herencia ligada a X.
En cuanto a la clasificación de los diagnósticos encontrados (Tabla 2), se observó que 13,2 % (n=20) correspondían a enfermedades cromosómicas (incluyendo sospecha de síndromes de microdeleción-microduplicación); 40,1 % (n=61) a enfermedades monogénicas o sospecha de estas; 22,4 % (n=28) a enfermedades de tipo multifactorial, 5,9 % (n=9) de tipo idiopático y 9,8 % (n=15) de tipo ambiental (Tabla 2). El 50,8 % de los diagnósticos o su sospecha fueron aportados por casos únicos, excepto en los diagnósticos de MPS III, Ellis-Van Creveld, X Frágil, Distrofia Cintura Miembro, Esclerosis Tuberosa y sospecha de Síndrome de Norrie.
Además se encontró un grupo de pacientes (12,5 %; n=19) con características clínicas heterogéneas tales como: neurorregresión, ataxia, corea, discapacidad cognitiva, manifestaciones sugestivas de enfermedad neuromuscular de origen metabólico, manifestaciones neurocutáneas, entre otras, que carecían de estudios previos útiles para el diagnóstico. En 6,6 % (n=10) de estos pacientes se considera la posibilidad de un error innato del metabolismo (EIM). Por tanto en los 19 pacientes se ordenaron estudios complementarios ya que la anamnesis y el examen físico no fueron suficientes -en estos casos difíciles- para orientar un diagnóstico específico.
Dentro de las enfermedades monogénicas, en el 5 % (n=8) se encontró diagnóstico clínico no confirmado de síndrome de X frágil. Un paciente (procedente de Tunja) tenía confirmación molecular de la enfermedad y por tanto se brindó el asesoramiento genético a esta familia.
Cabe anotar, que se encontraron 3,2 % (n=5) con cuadro compatible con Distrofia Cintura Miembro, siendo tres de ellos consanguíneos. El diagnóstico fue realizado clínicamente pues se solicitó prueba molecular confirmatoria la cual aún no había sido realizada previamente en estos pacientes.
Se encontró que del total de pacientes vistos, el 15,7 % (24 individuos) referían consanguinidad parental. De estos pacientes, se encontró consanguinidad de los padres en 21 individuos, y en tres pacientes la consanguinidad se presentó entre abuelos de ambas líneas parentales. En el 8,5 % de los casos (13 individuos) se desconoce el antecedente de consanguinidad, debido a que en su gran mayoría son pacientes en abandono social, bajo el cuidado del Estado, y en quienes no se dispone de datos de los padres, e incluso no hay datos pre o perinatales.
Por otro lado, se identificaron dos posibles agrupamientos de patologías raras con una frecuencia más alta de la esperada en la población general. Estas condiciones fueron el Síndrome de Ellis-Van Creveld (EVC) y la Mupolisacaridosis tipo III (MPS III).
Mucopolisacaridosis tipo III
Se identificaron 5 pacientes (3,2 %), cuatro de ellos procedentes de una misma familia con hallazgos clínicos compatibles con MPS III y excreción aumentada de Heparán Sulfato en orina. El Pedigree de 6 generaciones (datos no mostrados) de esta familia demostró ascendencia multifamiliar común y otros 6 individuos afectados. Al realizar el cálculo de frecuencias, se encontró una frecuencia más alta de la esperada en la población general (1:28 000), caracterizando así un agrupamiento de pacientes afectados por MPS III (Tabla 3).
Tabla 3 Incidencia de los agrupamientos de MPS III y EVC encontrados

* Se incluye otros familiares afectados, que no fueron evaluados clínicamente, pero fueron identificados en el árbol genealógico; ** Sobre la población de la región evaluada, es decir, 400 000 individuos.;***La prevalencia de 4,1 ha sido reportada únicamente en Países Bajos. En Colombia, la incidencia reportada es de 0,7/100 000 10.
Síndrome de Ellis Van Creveld (EVC)
Se identificaron 3 pacientes (2 %), dos de ellos consanguíneos, con hallazgos clínicos y paraclínicos compatibles con EVC. Al realizar el cálculo de frecuencias, se encontró una frecuencia de 0,7:100 000 (Tabla 3).
DISCUSIÓN
Con este tipo de actividades exploratorias sobre la situación de la patología genética en Boyacá, logramos encontrar un importante número de diagnósticos nuevos y se logró dar seguimiento a aquellos pacientes previamente valorados.
Se observó una gran heterogeneidad en los diagnósticos de síndromes monogénicos, siendo el 50,8 % aportados por casos únicos, excepto en los diagnósticos de MPS III, Ellis van Creveld, X Frágil, Distrofia Cintura Miembro, Esclerosis Tuberosa y sospecha de Síndrome de Norrie (Tabla 2). Esta heterogeneidad era de esperarse, pues como se documenta en la literatura, las enfermedades raras tienen una baja incidencia al ser consideradas por separado, pero esta incidencia cobra relevancia en conjunto 6,11.
El porcentaje de pacientes hijos de padres consanguíneos encontrado en nuestro estudio es de 15,7 %. En el mundo, las frecuencias reportadas de matrimonios consanguíneos son tan altas como 55,4 % en Afganistán, y tan bajas como 0,2 % en Australia, de acuerdo con los reportes del sitio web Global consanguinity (www.consang.net). En este mismo sitio, se ha reportado un porcentaje de consanguinidad en Colombia del 1,1 %, y sólo en la región antioqueña de 4,4 %, siendo en su gran mayoría uniones entre primos hermanos y en una menor proporción entre primos segundos.
En Colombia, los estudios de isonimia y haplotipificación se han realizado en su mayoría en Antioquia, donde se conoce que desde tiempos coloniales esta población se ha comportado como un aislado genético, cuyos polimorfismos del cromosoma Y son de descendencia europea, y el ADN mitocondrial es amerindio 12. No hay estudios sobre endogamia en la población de Boyacá, sin embargo, dado el elevado porcentaje de pacientes hijos de padres consanguíneos, es necesario determinar el porcentaje de endogamia en esta población, teniendo en cuenta que no todos los pacientes hijos de padres consanguíneos se encuentran afectados por enfermedad. Así como ha sido reportado en diferentes estudios 13, se infiere que la endogamia en determinada población se presenta por efecto de barreras geográficas, intereses económicos y otros aspectos socio-culturales que se encuentran fuera del propósito de este estudio 14.
Un aspecto interesante de la población analizada en este estudio fue el hallazgo de una frecuencia mayor de la esperada para dos enfermedades genéticas y un de ellas con un probable agrupamiento genético: síndrome de Ellis-Van Creveld 15 y Mucopolisacaridosis tipo III 16 (Tabla 3).
La mucopolisacaridosis tipo III, o Síndrome de Sanfilippo (OMIM 252900, 252920, 252930, 252940) es una enfermedad autosómica recesiva causada por la deficiencia en una de las 4 enzimas encargadas de la degradación del Heparán Sulfato 17. Clínicamente se caracteriza por presentar un desarrollo normal desde el nacimiento hasta la manifestación de síntomas, que se presenta entre los 2 y 6 años, con cambios comportamentales, regresión del neurodesarrollo, siendo éste más marcado a nivel del lenguaje, y alteraciones del ciclo circadiano 16. Al examen físico se evidencia facies tosca, con cejas prominentes y sinofris, cabello ralo y seco, y la gran mayoría presenta hipertricosis. No es común encontrar visceromegalias en estos pacientes 17.
El síndrome de Ellis-Van Creveld (OMIM 225500)es una displasia esquelética autosómica recesiva, vista en clusters en poblaciones aisladas como los Amish de la vieja orden de Pensilvania, caracterizada por acortamiento de miembros, tórax estrecho, polidactilia postaxial, y uñas y dientes displásicos. En el 60 % de los individuos puede haber anomalías cardíacas congénitas y su capacidad cognitiva es normal 18.
También llama la atención que se encontraron cinco casos de sospecha de Distrofia Cintura Miembro, tres de ellos consanguíneos, mientras los dos restantes procedían de familias diferentes. La confirmación del diagnóstico mediante la prueba molecular permitirá brindar datos confiables sobre la epidemiología de esta enfermedad en la región.
La Distrofia Cintura Miembro (OMIM 608099) es un grupo heterogéneo de trastornos hereditarios de debilidad muscular progresiva que afecta predominantemente los músculos de la cintura pélvica y escapular y que tienen patrones de herencia tanto autosómico dominante como recesivo. Los avances en su diagnóstico, incluyendo a tecnología de Secuenciación de Siguiente Generación (Next Generation Sequencing) expandió el número de formas reconocidas 19. La confirmación del diagnóstico ante un paciente con sospecha de enfermedad muscular es crucial para un adecuado manejo, pues el tratamiento con Creatina ha demostrado un aumento significativo de la fuerza muscular en pacientes con distrofinopatías 20, entidades que hacen parte del diagnóstico diferencial en pacientes con sospecha de distrofia cintura-miembro.
En cuanto a los errores innatos del metabolismo (EIM), se encontró una frecuencia importante de sospecha de estas enfermedades (6,6 %; n=10), incluyendo nuevos posibles casos de Enfermedad de Gaucher y Mucopolisacaridosis tipo II no neuronopático. Los EIM suelen tener presentaciones inespecíficas, como problemas de alimentación, regresión del neurodesarrollo, déficit cognitivo, ataxia, distonía, etc. lo cual, sumado al desconocimiento de estas entidades entre los profesionales de la salud, dificulta el acceso al servicio especializado que requieren estos pacientes para llegar al diagnóstico específico 21.
Ciertos EIM como la deficiencia de biotinidasa, fenilcetonuria, acidemias metilmalónica y propiónica, déficit de acil-CoA deshidrogenasa de cadena media (MCAD), defectos de la biosíntesis de Creatina y algunas enfermedades de depósito lisosomal (incluidas Gaucher y MPSII), disponen de tratamiento que puede cambiar de manera positiva el curso de la enfermedad, reduciendo la discapacidad asociada a la misma 22.
Por otra parte, observó un alto número de casos con sospecha de trastorno genómico, quienes no han sido estudiados de forma parcial. La Hibridación genómica comparativa por array (aCGH) es un examen de aplicación clínica disponible y según las guías del Colegio Americano de Genética Médica y el Colegio Americano de Pediatría este examen está indicado en pacientes con déficit cognitivo de origen no claro sumado a características dismórficas cuyo diagnóstico no puede ser resuelto por citogenética convencional. En estos casos, el aCGH aporta hasta un 19 % de rendimiento diagnóstico sobre el cariotipo bandeo G 23,24. Los síndromes de microdeleción-microduplicación deben ser confirmados mediante aCGH, ya que en las regiones con pérdida o ganancia de material genético pueden estar involucrados genes con predisposición a complicaciones específicas (por ejemplo ciertos tipos de cáncer, epilepsia, neuroregresión) lo cual tiene importancia en el manejo, seguimiento y pronóstico 25.
Observamos que el diagnóstico de muchas de estas patologías no es oportuno: 96 % de los pacientes están aún en proceso de confirmar su diagnóstico y la edad promedio es 14 años. Existen múltiples barreras de acceso que retrasan los procesos de manejo integral; la distribución de profesionales capacitados en el diagnóstico y manejo de estas condiciones es inequitativo ya que la mayoría suelen estar concentrados en centros metropolitanos con instituciones médicas de alto nivel de complejidad e instituciones de educación superior con programas de investigación biomédica. El entrenamiento en la identificación de pacientes con posibles enfermedades genéticas en las facultades de Medicina del país es limitado y la capacidad de detección en el primer nivel de atención en salud es heterogénea.
Lo anterior, ha sido observado en otros países en vías de desarrollo 8 y a su vez ha creado la necesidad de detección oportuna de dichas enfermedades puesto que el diagnóstico temprano puede ayudar a la instauración temprana de terapias que pueden modificar el curso de la enfermedad y disminuir la morbimortalidad asociada con dichas enfermedades. Estos hallazgos también refuerzan la necesidad de mejoramiento del acceso o un pronto diagnóstico de enfermedades genéticas ya que estas poblaciones demuestran tener un riesgo incrementado de patologías genéticas primordialmente de origen autosómico recesivo, con respecto a poblaciones donde estos fenómenos de aislamiento génico son infrecuentes 26.
En conclusión, la actividad de medicina genética comunitaria desarrollada para la detección de patología genética sindrómica en el departamento de Boyacá surgió como una iniciativa para mejorar el acceso a la consulta especializada de pacientes y familias con enfermedades genéticas y promover el conocimiento de las mismas en la región. La patología genética hace parte del grupo de enfermedades huérfanas y el nuevo marco legal de la salud en Colombia reconoce su importancia, así como el impacto positivo que tiene su detección oportuna, manejo y seguimiento en los indicadores de salud.
Esperamos que el apoyo brindado a los pacientes, familias y personal asistencial de los hospitales de los municipios participantes de esta actividad se traduzca a mediano y largo plazo en un diagnóstico más oportuno que permita el manejo, tratamiento específico cuando aplique y asesoramiento genético necesarios para una mejor calidad de vida, reducir la discapacidad y disminuir la morbimortalidad.

