











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkHypoparathyroidism, sensorineural deafness and renal disease (HDR) syndrome, also known as Barakat syndrome, is a hereditary autosomal dominant disease first described in 1977 by Barakat et al., when reporting the case of two male siblings with nephrotic syndrome, nerve deafness and hypoparathyroidism 1. In 1997, Hasewaga et al. 2 named it HDR syndrome, when they reported the case of a girl with the same clinical features and deletion of locus10p13 2.
A gene mutation has been identified on GATA3 gene, located on 10p15 loci; more than 50 GATA3 mutations have been associated with HDR syndrome, and haploin-sufficiency has been considered as the underlying mechanism 3-6. The syndrome has a wide range of penetrance and expressivity, which makes its diagnosis a challenge for medical health providers and in many cases, unfortunately, the condition is not diagnosed 7.
Clinical features include hypoparathyroidism at any age (probably the most specific symptom), and calcium levels variations, from normal to very low levels, which may lead to tetany and seizures. Undetectable or very low serum PTH levels is one of the main features of HDR syndrome 6,7, and an early sensorineural deafness onset (usually bilateral) is a common characteristic and the most penetrant6,7. Renal abnormalities can be unilateral or bilateral and may vary from minor to severe 6-8. Genitourinary malformations have also been reported a few times, and have been associated with GATA3 haploinsufficiency 9-11.
Currently, HDR syndrome prevalence is unknown, for less than 120 cases have been reported since Barakat et al. described the first report 1,2,6,7,12. Taking this into account, this would be the first case to be reported in Latin America, and as authors we believe publishing this report will help to raise awareness on the occurrence and existence of rare diseases in our regional context.
Case presentation
A 36-year-old Colombian female with a diagnosis of parathyroid gland agenesis and hypocalcaemia was referred from the Endocrinology Service to the Outpatient Medical Genetics Service. The patient was asymptomatic and according to her medical history, she had left renal agenesis and bicornuate uterus, conditions that were found out incidentally after an ultrasound was made. Also, she had been diagnosed with hypoparathyroidism and sensorineural hearing loss, which was assumed to be secondary to a measles infection in her childhood. In addition, her sister had been diagnosed with hypocalcaemia, osteoporosis and hearing loss, and her mother, hypocalcaemia and osteoporosis (Figure 1). The patient had been taking 3600 mg per day of calcium carbonate and 0.5 mcg per day of calcitriol (1, 25-dihydroxyvitamin D3).
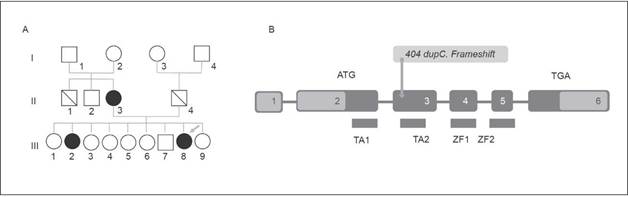
Figure 1 A) Pedigree. The arrow points at the patient described (III-8), members of her family which display at least one of the three typical findings of the HDR Syndrome are in the filled dark Circles; III-2 had hypocalcaemia with hearing loss and II-3 had as unique finding a hypocalcaemia. B) Schematic Illustration of GATA3 Gene with yellow arrow locating patient's mutation (404 dupC. Frameshift). (Own elaboration)
After the physical examination was made, the following characteristics were observed: short stature, short neck, micrognathia, proptosis, posteriorly rotated ears, fourth toe clinodactyly and presence of bilateral hearing aids (Figure 2). Her vital signs were normal, she was 1.49 mts and weighed64.5 kg (BMI: 32.91). Her relatives had similar facies and phenotype, a fact that drew the attention of the authors of this article.
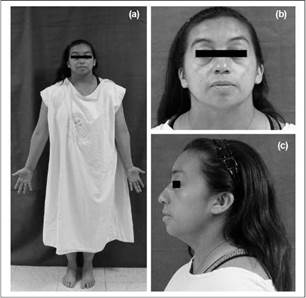
Figure 2 Patient Photographs. A) Patient standing in anatomical position note short stature and short neck B) Anterior image of patient's face showing short neck and posteriorly rotated ears C) Lateral image of patient's face showing Hearing aid, mild micrognathia and posteriorly rotated ear. (Own elaboration)
The first diagnostic impression was that the patient had HDR syndrome, since laboratory tests reported a low PTH level and a slightly low serum calcium level, somehow serum calcium level was normal in further studies. Based on evoked potentials, a neurosensory bilateral involvement with a peripheral lesion was confirmed, while left renal agenesis, right simple renal cysts, and bicornuate uterus were confirmed through and abdominal ultrasonography. In addition, an MRI scan was performed, in which complete septate uterus was observed. Ovaries were normal. The karyotype was normal (46 XX), and through a bidirectional ng sequencing of GATA 3 gene, performed in an AB13130 automatic sequencer, a pathological mutation was found: c.404dupC (p. Ala136 GlyfsTER 167) (Figure 1).
DISCUSSION
A GATA3 gene mutation was found in the patient (Figure 1), the mutated gene showed a cytosine duplication at position 404, which caused a glycine to be replaced by an alanine at position 136 of the protein, resulting in a frame shift producing a terminal codon that ended the protein prematurely. This mutation disrupts TA2 (transactivating-2) domain and two zinc fingers (ZnF1 and ZnF1), which causes the same effect described by Adachi et al. in their report of a Japanese girl and her father 12.
The GATA3 gene expression can be detected from the beginning of the 4th gestational week in humans13. GATA3 transcripts are mainly observed in parathyroid glands, inner ear and kidneys, which correlates with the phenotype of patients with HDR syndrome, nevertheless the GATA3 expression can also be detected in multiple tissues and organs like the developing central nervous system (CNS), the liver, the foregut and the eyes, but these organs seem to be less prone to the haploinsufficiency of GATA3 since they are not generally affected in patients with this syndrome 13.
On the other hand, some reports describing patients with HDR syndrome and who additionally developed genitourinary tract abnormalities (as it happened in the case reported here) suggest that the underlying mechanism that causes the syndrome is the same triggering in the development of genitourinary abnormalities, since GATA3 seems to be involved in uterine development9,10,14.
Orphan diseases (OD) such as HDR syndrome have an estimated frequency that ranges from 37.3 and 52.8 per 1000 people in Colombia 15. The prevalence of this genetic disease is increasing and it has been concluded that it is underdiagnosed, which leads to inaccurate reports of the syndrome in the country's databases and, therefore, a huge impact in terms of public health caused by the amount of years lost to disability (YLDS) in these patients 15,16.
Unawareness on this disease may lead to underdiagnosis in Latin-American countries. The diversity found in the Colombian population makes it necessary to suspect genetic diseases traditionally reported in other countries in patients presenting typical symptoms. HDR syndrome must be suspected regardless of ethnicity, and thus the patient must be referred to a human genetics specialist as soon as possible to provide an early diagnosis and counseling on the condition•

