










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkPerspectivas en Nutrición Humana
Print version ISSN 0124-4108
Perspect Nut Hum vol.12 no.2 Medellín July/Dec. 2010
ARTÍCULO DE REVISIÓN
Hígado graso no alcohólico: ¿un componente inflamatorio del síndrome metabólico?
Is a non-alcoholic fatty liver an inflammatory component of the metabolic syndrome?
Johan Almarza1
1Centro de Investigaciones Endocrino-Metabólicas Dr. Félix Gómez. Maracaibo- Venezuela. johanalmarza@gmail.com. Premio Federación Latinoamericana de Nutrición Clínica y Metabolismo (FELANPE)
Artículo recibido: 14 de octubre de 2010 Aceptado: 6 de diciembre de 2010
Como citar este artículo: Almarza J. Hígado graso no alcohólico: ¿un componente inflamatorio del síndrome metabólico? Perspect Nutr Humana. 2010;12:163-175.
Resumen
Objetivo: integrar los componentes que relacionan la insulinorresistencia en el desarrollo de hígado graso no alcohólico y los mecanismos moleculares implicados en la fisiopatología, los métodos diagnósticos y las implicaciones clínicas. Materiales y métodos: se realizó una búsqueda sistemática de artículos desde 1990 a 2010 en el periodo comprendido entre octubre de 2009 a diciembre de 2010, seleccionados de bases de datos internacionales PubMed y en los servicios ScienceDirect y Ovid; la búsqueda de información relacionada con América Latina, incluyendo Colombia, y Venezuela, se efectuó en Lilacs y Scielo. Para la elección de los términos se utilizó el Medical Subject Headings (Mesh) y los vocabularios controlados de cada uno de los servicios consultados. Resultados y conclusiones: los mediadores inflamatorios (adipoquinas), oxidativos (especies reactivas de oxígeno) y metabólicos (ácidos grasos libres) responsables de la historia natural del SM están relacionados con el desarrollo del Hígado Graso no Alcohólico (HGNA), por lo tanto la identificación de este componente patológico, mediante lo métodos bioquímicos y ecográficos pertinentes, es de suma importancia. La pérdida gradual de peso, el aumento de la actividad física más las modificaciones dietarias que aumentan la expresión de enzimas peroxisomales de la beta oxidación de ácidos grasos, y previenen su posterior acumulación citoplasmática.
Palabras clave: hígado graso, síndrome metabólico, estrés oxidativo.
Abstract
Objective: Integrate components that correlate insulin-resistance in the development of non-alcoholic fatty liver and the molecular mechanisms involved in the pathophysiology, diagnostic methods and clinical implications. Methods and materials: A systematic search was conducted of articles from 1990 to 2010, comprised primarily of a period from October 2009 to December 2010, using international data bases like PubMed, ScienceDirect and Ovid. The research for information related to Latin America, including Colombia and Venezuela, was done in Lilacs and Scielo. Medical Subject Headings (Mesh) and each one of the said services were used in determining choice of terms. Results and conclusions: Inflammatory mediators (adipokines), oxidative (reactive oxygen species) and metabolic (free fatty acids) responsible for the natural history of the SM are relative to the development of the non-alcoholic fatty liver (HGNA); therefore the identification of this pathological component through the biochemical methods and relevant ultrasound, is of the utmost importance. Gradual weight loss, increased physical activity, dietary modifications increase the expression of enzymes peroxisomes of the beta oxidation of fatty acids, and prevent their subsequent cytoplasmic accumulation.
Key words: fatty liver, metabolic syndrome, oxidative stress.
INTRODUCCIÓN
El síndrome metabólico (SM) se define como un conjunto de patologías asociadas con insulinorresistencia (IR), que pueden contribuir de manera sinérgica o aislada al desarrollo de Diabetes tipo 2 (DT2), enfermedad cardiovascular (ECV) o a ambas (1-3). La causa del SM se desconoce. Su fisiopatología es extremadamente compleja y solo ha sido dilucidada una parte de ella. La mayoría de los pacientes son obesos, sedentarios y tienen cierto grado de resistencia a la insulina. Este último factor desempeña un papel central en la génesis de este SM (1). Por otra parte, algunas patologías auntoinmunes, cáncer e hígado graso no alcohólico, están estrechamente relacionadas con el SM; la valoración y descarte de estas condiciones es básicamente ignorada al momento de examinar al paciente.
Se define al hígado graso no alcohólico (HGNA) como la infiltración de grasa macrovesicular en el hígado, que excede de 5 a 10% del peso del mismo (4-5). Es una entidad patológica que abarca un espectro de alteraciones que van desde la infiltración hasta la fibrosis, e incluso cirrosis, con las complicaciones a que esto lleva, como hipertensión portal y carcinoma hepatocelular, hallazgos histopatológicos descritos por primera vez por Ludwig (6-10). Los hallazgos histológicos en el HGNA son indistinguibles de los que se observan en la esteatohepatitis alcohólica, por lo cual se ha aceptado que para su diagnóstico se requiere que los pacientes no ingieran bebidas alcohólicas o bien no ingieran más de tres onzas semanales de bebidas destiladas a la semana (12). El HGNA, considerado la patológica hepática más común en países occidentales, afecta entre 10 y 24% de la población general, porcentaje que oscila entre 58 y 74% en personas obesas (13). Los mediadores inflamatorios y hormonales implicados en el SM actúan en sinergia en el desarrollo del HGNA; común denominador en pacientes obesos e insulinorresistentes (12). Por las premisas antes descritas, el propósito de esta revisión es hacer una sinergia en los factores hormonales e inflamatorios involucrados en la obesidad y SM y su papel en el desarrollo del HGNA.
Papel de la obesidad en el desarrollo de HGNA
La obesidad se define como una condición patológica que se caracteriza por el incremento de la masa corporal a expensas del compartimiento graso. El diagnóstico de la misma se puede hacer basándose en el índice de masa corporal (IMC) mayor o igual a 30Kg/m2 en la población adulta o superior al percentil 90 de los valores del HNANES III en la población pediátrica (10). La obesidad se caracteriza por presentar una serie de comorbilidades (hipertensión, dislipidemias y apnea del sueño), las cuales alteran la calidad de vida del paciente y desencadenan otras patologías crónicas como diabetes y cáncer, entre otras (11). La obesidad es considerada multifactorial, los elementos involucrados directamente sobre el desarrollo de la misma son hábitos alimentarios inadecuados, sedentarismo y factores psicológicos (11); sin embargo, en los últimos años se ha reconocido que la interacción del genotipo y el estilo de vida pueden contribuir dramáticamente a la incidencia y prevalencia de la obesidad (12). Muchos de los mediadores fisiopatológicos de esta enfermedad desencadenan reacciones que alteran el funcionamiento hepático; la oferta de ácidos grasos provenientes del tejido adiposo incrementan el proceso de filtración (o infiltración) grasa y fibrosis del tejido hepático (11-15).
Síndrome metabólico e hígado graso no alcohólico
Una de las condiciones desencadenantes del desarrollo del hígado graso es el SM, cuyo detonante fisiopatológico es la obesidad, en especial la de distribución central (10). De esta manera, la historia natural del hígado graso en individuos obesos está sincronizada con la patogénesis de la obesidad, el SM y la diabetes mellitus tipo 2 debido a un efecto sinérgico entre los mediadores proinflamatorios característicos de estas condiciones. Entre los mediadores inflamatorios y humorales implicados con la fisiopatología de la esteatosis hepática están el TNF alfa y la Interleucina-6, secretados por el tejido adiposo (adipoquinas) (15-16). Además de de lo anterior se destaca la hiperleptinemia a causa de la leptinorresistencia, junto con el descenso de adiponectina, entre otras adipoquinas que son afectadas por el fenómeno de hipertrofia e insulinorresistencia causados por el tejido adiposo (4-11, 15-16). Estos mediadores fisiopatológicos desencadenan reacciones que alteran el funcionamiento hepático, por la oferta de ácidos grasos provenientes del tejido adiposo, incrementando el proceso de filtración grasa y fibrosis del tejido hepático; sin embargo, en la obesidad, la insulinorresistencia desempeña un rol central en el desarrollo de la misma y al mismo tiempo en la patogénesis de la esteatosis hepática, lo cual puede desencadenar problemas más serios como fibrosis e insuficiencia hepática (4-11).
Historia natural del hígado graso: papel de la insulinorresistencia y el estrés oxidatívo
El hígado graso no alcohólico se encuentra asociado fuertemente con un estado de insulinorresistencia del tejido adiposo. Esta condición promueve la oferta de ácidos grasos cuyo destino es la formación de lipoproteínas de baja densidad (VLDL), beta oxidación peroxisomal y activación de los receptores PPAR (receptores activadores de la proliferación de los peroxisomas) a nivel nuclear para inducir la expresión de las enzimas claves de la beta oxidación (figura 1A). Por otra parte, el hígado es uno de los órganos afectados por el estado de insulinorresistencia, lo que disminuye el metabolismo lipídico causando el acúmulo de triacilglicéridos, desencadenando procesos inflamatorios que conllevan al proceso de fibrosis hepática (17-21). La mitocondria del hepatocito es el organelo más afectado por esta condición, la sobreoferta de ácidos grasos promueve la formación de especies reactivas de oxígeno (ROS), contribuyendo al proceso de apoptosis y estímulo del proceso de fibrosis (citoqueratosis), conllevando al remplazo de tejido sano por tejido fibroso no funcional (figura 1B) (18).
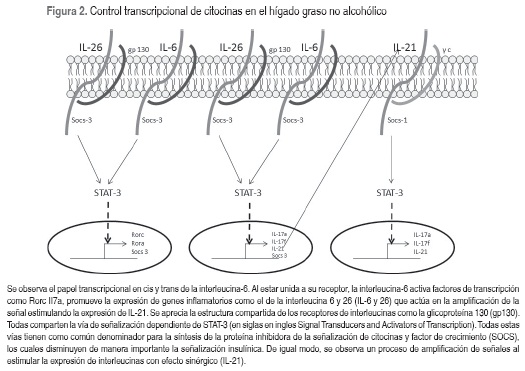
Estas evidencias sugieren que la infiltración de grasa hepática es directamente proporcional a la insulinorresistencia y la grasa visceral (grasa intrabdominal). La sobreoferta de ácidos grasos, característica en estos pacientes, contribuye al incremento de diacilglicéridos, los cuales activan la protein kinasa C (PKC), la cual fosforila en residuos de serina y treonina del substrato del receptor insulínico 1 (IRS-1) disminuyendo la señalización insulínica (22-25). En estado de insulinorresistencia se incrementa la expresión de las enzimas lipogénicas vía proteína de unión de esteroles (SREBP-1); lo anterior conduce al acúmulo de grasa en el hepatocito. Por otro lado el flujo de glucosa en estados de intolerancia a la glucosa durante la insulinorresistencia, conduce a la activación de los receptores activados por carbohidratos, los cuales, de igual modo, incrementan la expresión de las enzimas lipogénicas contribuyendo al acúmulo de grasa (20-23). Otro factor importante involucrado en la infiltración de grasa hepática es la inactivación del factor de transcripción Foxa 2, el cual está involucrado en la transcripción de genes de la beta oxidación de ácidos grasos. En condiciones normales es fosforilado y activado por el IRS-1. Estando el IRS fosforilado en residuos de serina y treonina, la activación del Foxa 2 no se lleva a cabo, por lo tanto converge en la inactivación de la cascada insulínica (20-25).Por otra parte, en la obesidad e insulinorresistencia se rompe el balance de las adipoquinas por disminución de las concentraciones de adiponectina, la cual en condiciones fisiológicas incrementa la actividad de la AMP quinasa cuya función es favorecer la β-oxidación de ácidos grasos en el hígado. También, la expresión de enzimas prooxidantes contribuye a la fibrosis mediante el incremento de la expresión de mediadores inflamatorios (figura 2) (24).
Alteraciones metabólicas relacionadas con la fisiopatología del hígado graso
Metabolismo glucídico: el hígado y el páncreas son las glándulas principales, sensoras y reguladoras de las concentraciones plasmáticas de glucosa. En estados de insulinorresistencia y fibrosis hepática esta función se ve abolida, lo que conlleva a alteraciones de la glicemia por la incapacidad del órgano de responder a la insulina o a las hormonas contrarreguladoras respectivamente (12).
Metabolismo lipídico: el balance entre la síntesis y degradación de lípidos es una de las funciones fundamentales del hepatocito. Las fuentes corporales principales de lípidos son la síntesis de novo mediante la vía lipogénica en hígado, debido al exceso de glucosa que satura las reservas de glucógeno, e incremento de la tasa glucolítica, oferta de ácidos grasos provenientes de los quilomicrones en estado postprandial y del tejido adiposo (figura 3A). En estado de insulinorresistencia el balance se ve afectado debido a la producción de VLDL, inhibición de las enzimas lipogénicas y lipolílitas contribuyendo a un estado de hipertriacilgliceridemia y fibrosis hepática (12, 24-29). Por otra parte, en estados de IR la producción de la apoproteína B100 se ve afectada desviando la síntesis de VLDL hacia la acumulación citoesquelética de ácidos grasos, aumentando la actividad de los citocromos P450 y, de este modo, la producción de radicales libres (figura 3B).
Metabolismo proteico: en estados de malnutrición proteica (Kwashiorkor) existe una deficiencia de aporte de aminoácidos necesarios para la síntesis de las apoproteínas de las lipoproteínas de origen hepático (VLDL, HDL). Bajo estas condiciones ocurre el almacenamiento de lípidos dentro del hepatocito, lo que conlleva a la infiltración grasa y, consecuentemente, a las esteatosis y fibrosis hepática. Lo anterior explica la alta prevalencia de insuficiencia hepática en este grupo poblacional. Por otro lado, en el estado post operatorio de cirugías bariátricas ocurre depleción de nutrientes, contribuyendo a la deficiencia proteica y a la sobreoferta de ácidos grasos provenientes del tejido adiposo, originando una vía directa para la infiltración grasa (12).
Papel de las citocinas involucradas en el proceso inflamatorio
Durante procesos de inflamación sistémica, como SM , y en estadios de respuesta metabólica al estrés, se incrementa la síntesis y la secreción de citocinas proinflamatorias, como las interleucinas 1 y 6 (IL-1, IL-6), factor de TNF-a necrosis tumoral. Este ambiente inmunológico propicia un estado de insulinorresistencia y la síntesis de tejido no funcional en hígado, favoreciendo la fibrosis. Dichas citocinas tienen un mecanismo de acción específico que en convergencia ayudan al proceso fisiopatológico del hígado graso.
Interleucina-6 (IL-6)
Considerada como marcador inmunológico en procesos inflamatorios, esta citocina puede ser secretada de igual forma en el tejido adiposo como en las células del sistema inmune (macrófagos, linfocitos B, entre otros). Se une a su receptor, que se dimeriza, y mediante la vía de la janus kinasas –factores de transcripción traductores de señales (vía jak-STAT) promueve la transcripción de genes involucrados en la amplificación y redundancia de la respuesta inflamatoria. (Figura 2) (30-33). Siendo la obesidad un factor de riesgo para el desarrollo del hígado graso es importante considerar la relación entre las concentraciones de IL-6 y la gravedad de la lesión hepática (30-33).
Papel del factor de necrosis tumoral alfa) TNF-a
El TNF-a participa en la homeostasis del hepatocito controlando su tasa de supervivencia y muerte. Esta citocina se encuentra en concentraciones altas durante el proceso de infiltración grasa y es secretada de forma endocrina en el tejido adiposo visceral, lo que convierte a la grasa androide como un factor de riesgo para altas concentraciones de la citocina. Cuando se rompe el balance en las concentraciones de TNF-a ocurre un desbalance homeostático hepático conduciendo al carcinoma o la apoptosis (fibrosis). Por otro lado, por vía de las ceramidas y generación de especies reactivas de oxigeno puede originarse la muerte programada y necrosis del hepatocito (vía de la mitocondria) (34). Cuando se une el TNF al receptor ocurre una trimerización para la formación del complejo 1, compuesto por el dominio de muerte del receptor trimerizado, proteínas traf (factor asociado al receptor de TNF), que luego por una cascada de quinasas fosforilan e inactivan al inhibidor del factor nuclear kappa B para ser reconocido y posteriormente ubiquitinado, liberando al factor de transcripción, ejerciendo su función transcripcional. De igual modo el complejo 1 puede fosforilar a las proteínas activadas por mitógenos (MAP p38) y a la jun quinasas (JNK), estimulando la transcripción de genes pro inflamatorios. Por otro lado puede formarse un complejo II al cual se le une una procaspasa 8, iniciándose el proceso de apoptosis (34). La caspasa 8 provoca activación de la proteína Bid (Bcl-2 Interacting Domain), que se encarga del reclutamiento de las Bak y Bax (proteínas de la familia Bcl proapoptóticas) para la formación del poro en la membrana mitocondrial, de modo que pueda ser más permeable y pueda salir de la misma el citocromo c, formándose el complejo apoptosoma con la Apaf 1 (Factor de Activación Apoptotic Proteína-1), procaspasa 9 y ATP, este último se encarga de la activación de la caspasa 3 ejecutando la apoptosis. El Bid activado puede abrir los lisosomas liberando las catepsinas, las cuales por vía caspasa 2 incrementan la permeabilidad de la membrana mitocondrial a los radicales libres y citocromos. Cuando la producción de radicales libres empieza a ser citotóxica ocurre una muerte desprogramada y necrosis descontrolada del hepatocito (33).
Valoración clínica
Como suele ocurrir en casi toda hepatopatía crónica, el curso clínico es silente, y en el caso del HGNA, este es usualmente descubierto durante la evaluación del paciente con niveles elevados de aminotransferasas. La presencia de íctericia o signos de hepatopatía crónica, tales como ascitis, várices esofágicas o encefalopatía hepática, se ven con poca frecuencia e invariablemente indican enfermedad severa (4). Es importante realizar un diagnóstico diferencial y aislar otras causas de elevación de las aminotransferasas como hepatitis viral B o C, hemocromatosis, hepatitis autoinmune, deficiencia de alfa-1 antitripsina, enfermedad de Wilson o procesos obstructivos biliares. La obtención de las respectivas pruebas de descarte dependerá de las características particulares del paciente (antropometría, antecedentes personales y familiares) y del juicio clínico del médico tratante (28-29). La relación AST/ALT (aspartato aminotransferasa/alanino aminotransferasa) es generalmente <1, aunque cuando es >1 puede indicar presencia de fibrosis según algunos estudios. Sin embargo, también puede indicar enfermedad por alcohol, debido a que la AST es de producción mitocondrial, que se afecta mucho por el metabolismo del etanol, siendo un marcador de hepatitis alcohólica sobre todo si la relación es > de 2. Pese a esto, es importante señalar que el grado de elevación de ALT no tiene correlación con el grado de fibrosis hepática. Puede haber elevación de la fosfatasa alcalina y de la gamma glutamil transpeptidasa en grados variables, aunque dichas enzimas no son usualmente el parámetro de seguimiento más importante en HGNA (12-18).
Sin embargo, aun con los parámetros bioquímicos dentro de los rangos normales, si el paciente se caracteriza por tener los componentes considerados de diagnóstico de SM más una ecografía hepatobiliar que indique esteatosis según el criterio del ecografista, es importante la intervención terapéutica inmediata. Cuando se presentan valores de transaminasas dentro de los parámetros normales y de esteatosis según los criterios ecográficos, se puede sospechar de HGNA, que probablemente ha llevado años en desarrollarse, por lo general, en casos agudos es que se observa alteración de las enzimas.
Los criterios ultrasonográficos más comúnmente utilizados para diagnosticar esteatosis son: hígado hiperecogénico (''brillo'' sonográfico), ecodensidad comparada con los riñones y borramiento de estructuras vasculares. La tomografía axial computarizada (TAC) también puede ser de utilidad para sospechar hígado graso. Si la densidad tomográfica del hígado es mayor que la del bazo, la presencia de esteatosis puede descartarse con relativa precisión. La resonancia magnética nuclear (RMN) ha sido también investigada en este contexto, aunque con evidentes desventajas en cuanto a costo en comparación con las otras modalidades imagenológicas. Más aún, la sensibilidad de la ecografía es mayor para la detección de esteatosis en comparación con TAC, aunque en casos de esteatosis focal o no uniforme la TAC posee mayor precisión (12-18).
La fibrosis hepática severa es indistinguible de la esteatosis simple por modalidades imagenológicas, por lo cual la biopsia hepática es la única manera de hacer la distinción entre HGNA simple y fibrosis hepática. Ninguna de dichas modalidades imagenológicas es capaz de predecir la presencia de balonamiento celular, cuerpos de Mallory o fibrosis. La severidad de la esteatosis es determinada radiológicamente solo cuando había infiltración grasa moderada a severa (> 33%), documentada por biopsia hepática (12).
La realización de la biopsia hepática cuando se sospecha alguna de las formas del espectro de HGNA es aún materia de controversia. Por un lado, se argumenta que es un procedimiento invasivo, con riesgos potenciales y costosos, aparte de no ofrecer terapia específica de confirmarse HGNA simple o EHNA. Sin embargo, permite establecer el diagnóstico preciso y el pronóstico según el grado histológico. La evaluación clínica y de laboratorio para el diagnóstico de EHNA, tiene un valor predictivo positivo bajo (56%) (10). Por otro lado, la utilidad de la biopsia hepática en la práctica clínica en un escenario de aminotransferasas elevadas sin causa detectable ha sido recientemente evaluada. La esteatosis observada en las biopsias de pacientes con HGNA es generalmente de tipo macrovesicular, aunque en algunos casos se observa esteatosis microvesicular. Puede verse en el microscopio necrosis focal en grado variable en el lobulillo hepático, constituida por diversas células, entre ellas linfocitos, células de Kupffer, o polimorfonucleares (5, 7-10). Es importante destacar que cuando existe infiltrado predominantemente portal esto no es compatible con el diagnóstico de fibrosis hepática y se debe descartar otro tipo de patología como hepatitis crónica viral o autoinmune. Tal como se ha descrito, en la hepatitis alcohólica, en EHNA ocurre daño al citoesqueleto produciendo balonamiento del hepatocito y presencia de cuerpos de Mallory (cuerpos hialinos acidófilos) y su presencia no es tan frecuentemente observada como lo es en el caso de la hepatitis alcohólica (5).
Los estadios más tempranos de fibrosis en HGNA se observan en los espacios perisinusoidales con localización preferencial en la zona perivenular o zona 3. Estadios más avanzados muestran un mayor grado de fibrosis, pudiéndose ver puentes entre zona 3 y espacios porta. Por último, un grupo de pacientes puede presentar cirrosis, aunque en esta etapa histológica las características típicas de EHNA (esteatosis, inflamación, cuerpos de Mallory) pueden desaparecer (5).
Tratamiento del hígado graso no alcohólico
Considerando los diversos procesos fisiopatológicos involucrados en el desarrollo y progreso del HGNA, las modalidades terapéuticas comparten esta misma diversidad, con distintos resultados. Las estrategias de tratamiento comparten puntos primordiales, principalmente el contrarrestar los efectos del SM de resistencia a la insulina como obesidad, diabetes y dislipidemia; los medios para lograr este objetivo han sido abordados desde diversas disciplinas. Otro elemento que se debe considerar en el tratamiento del HGNA es la divergencia de respuesta al tratamiento entre los modelos experimentales, animales y humanos, los cuales no siempre guardan una adecuada concordancia.
Reducción de peso
La reducción de peso ha demostrado mejorar la sensibilidad a la insulina, por lo que el HGNA puede mostrar mejoría con un programa de reducción de peso. Existen catorce estudios publicados donde se demuestran los resultados del control del peso por medio de la dieta. Estos estudios presentan diversas deficiencias metodológicas, como poco control del estilo de vida de los pacientes estudiados, automedicación y muchos se realizan bypass gástrico, lo que limita la posibilidad de afirmar contundentemente los efectos del control dietético en el HGNA. Hasta este momento, se ha demostrado que el control dietético ha mejorado las variables bioquímicas de los pacientes con HGNA, aunque no en todos los casos se pudo corroborar si esta mejora bioquímica se acompañaba de cambios morfológicos demostrados por biopsia.
Los cambios histológicos son más evidentes después de una disminución de 11 a 20 kg de peso durante un año, es notable que en casos de disminución de peso de forma abrupta pueda existir disminución del daño morfológico documentado por histología sin que muestre una mejoría en marcadores bioquímicos. Estos cambios tanto bioquímicos como histológicos se consiguen en condiciones controladas en las que el manejo dietético se basa en dietas muy bajas en calorías (500 calorías/día). Se requiere al menos una disminución de 10% del peso corporal para conseguir modificaciones en las variables bioquímicas (43). Sin embargo, no se recomiendan cambios drásticos en el peso corporal porque causan un incremento en los niveles circulantes de ácidos grasos libres derivados de la movilización del tejido adiposo, incrementando sus niveles intrahepáticos favoreciendo el stress oxidativo, peroxidación de lípidos e inducción de citocinas que, en conjunto, empeoran el daño hepático (43). Se recomienda que el objetivo inicial del control del peso sea una pérdida de 10% en un periodo de seis meses, es decir, una pérdida aproximada de 450 a 900 g por semana.
En aquellos pacientes con HGNA pero sin obesidad se debe hacer mayor énfasis en el cambio de los componentes de la dieta y no así en la disminución de la ingesta calórica, además de favorecer la actividad física pues, al parecer, estos pacientes son candidatos a la terapia farmacológica. En el caso de los pacientes con HGNA, diabetes mellitus y/o dislipidemia, la primera medida terapéutica es el adecuado control del peso, que mejora la glicemia y los lípidos, aunque no siempre se acompaña de mejoría en la condición hepática, por lo tanto se deben tratar como entidades independientes aunque tienen la misma causa.
Tratamiento farmacológico
El uso de rosiglitazona y pioglitazona ha mostrado disminución de los niveles de enzimas hepáticas, que van de forma paralela a la disminución de la resistencia a la insulina, sin embargo, se debe monitorear el peso y composición corporal por la aparición de edema. Otro de los sensibilizadores de la insulina es la metformina, la cual en modelos animales ha ocasionado disminución de la hepatomegalia y el grado de esteatosis, así como normalización de los niveles de aminotransferasas. Resultados similares se han observado en humanos; sin embargo hay que mantener cautela durante su dosificación cuando las enzimas de funcionamiento hepático estén elevadas. No obstante, la rosiglitazona recientemente fue retirada del mercado por sus efectos pleiotrópicos. El uso de diversos antioxidantes ha mostrado cierta utilidad en el manejo del HGNA, dentro de este grupo de fármacos se incluyen: vitamina E, vitamina C, betaína, N-acetilcisteína y depleción de hierro (44-52).
CONCLUSIÓN
Dado que la esteatosis hepática está íntimamente relacionada con la fisiopatología de la obesidad y el SM, los mediadores inflamatorios e inmunológicos entre este último y la esteatosis hepática se presentan de manera paralela. Desde etapas tempranas del desarrollo de la vida es posible la toma de medidas preventivas cuyo objetivo sea la prevención del SM y sus patologías asociadas. El hígado graso no alcohólico todavía no es considerado por algún organismo oficial como uno de los componentes del SM ; sin embargo, al ser condiciones relacionadas, es necesario la consideración de esta patología en la valoración del paciente obeso y con SM , y el abordaje terapéutico inmediato. En la práctica clínica es poco considerado este aspecto, pero al analizar las interrelaciones inflamatorias se puede concluir que la valoración de los parámetros hepáticos, tanto bioquímicos y ecográficos, sería de gran utilidad para el manejo del paciente obeso y con SM. De acuerdo con esta revisión en el manejo de los pacientes obesos es importante el monitoreo de las pruebas de función hepática, que justifica, aún más, la vigilancia de la función hepatobiliar en este grupo poblacional considerándolo como parte de los componentes fisiopatológicos del SM en la mayoría de los casos.
REFERENCIAS
1. Cornier M, Dabelea D, Hernández T, Lindstrom R, Steig A, Stob N, et al. The metabolic syndrome. Endocrinol Rev. 2008;29:777-822. [ Links ]
2. Bagry H. Metabolic syndrome and insulin resistance. Anesthesiology. 2008;108:506-23. [ Links ]
3. Eckel R. The metabolic SM. Lancet. 2005;365:1415-28. [ Links ]
4. Roden. Mechanisms of disease:hepatic steatosis in type 2 diabetes-pathogenesis and clinical relevance. Nat Clin Pract Endocrinol Metab. 2006;2:335-48. [ Links ]
5. Mendez-Sánchez N, Chavez N, Uribe M. Hígado graso no alcohólico: nuevos conceptos. Investigación Clínica. 2004;56:72-82. [ Links ]
6. Ludwig J, McGill DB, Lindor KD. Review: nonalcoholic steatohepatitis. J Gastro Hepatol. 1997;12:398-03. [ Links ]
7. Castera L. Non-invasive diagnosis of steatosis and fibrosis. Diabetes Metab. 2008;34:674-79. [ Links ]
8. Mehta S, Lau B, Afdhal NH, Thomas DL. Exceeding the limits of liver histology markers. J Hepatol. 2009;50:36-41. [ Links ]
9. Poynard T, Muntenau M, Morra R, Ngo Y, Imbert-Bismut F, Thabut D. Methodological aspects for the interpretation of liver fibrosis non-invasive biomarkers: a 2008 update. Gastroenterol Clin Biol. 2008;32:8-21. [ Links ]
10. Bedossa P, Carrat F. Liver biopsy: the best, not the gold standard. J Hepatol. 2009;50:1-3. [ Links ]
11. Lemoine S. Definition and natural history of metabolic steatosis clinical aspects of NAFLD, NASH and cirrosis. Diabetes Metab. 2008;34:634-7. [ Links ]
12. Brent A. Neuschwander-Tetri. Fatty liver and the metabolic SM. Curr Opin Gastroenterol. 2007;23:193-8. [ Links ]
13. Tordjmana J, Clémenta M, Guerre-Milloa K. Adipose tissue inflammation and liver pathology in human obesity. Diabetes Metab. 2008;34 658-63. [ Links ]
14. Lafontan M, Girard J. Impact of visceral adipose tissue on liver metabolism. Part I: Heterogeneity of adipose tissue and functional properties of visceral adipose tissue. Diabetes Metab. 2008;34:317-27. [ Links ]
15. Girard J, Lafontan M. Impact of visceral adipose tissue on liver metabolism and insulin resistance. Part II: Visceral adipose tissue production and liver metabolism. Diabetes Metab. 2008;34:439-45. [ Links ]
16. Mutch DM, Clément K. Genetics of human obesity. Best Pract Res Clin Endocrinol Metab. 2006;20:647-64. [ Links ]
17. Tillotson J. America's obesity: conflicting public policies, industrial economic development, and unintended human consequences. Annu Rev Nutr. 2004;24:617-43. [ Links ]
18. Mantena S. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol and obesity-induced fatty liver diseases. Free Radic Biol Med. 2008;44:1259-72. [ Links ]
19. Perlemuter G, Bigorgne A, Cassard-Doulcier A, Naveau S. Nonalcoholic fatty liver disease: from pathogenesis to patient care. Nat Clin Pract Endocrinol Metab. 2007;3:458-69. [ Links ]
20. Caldwell SH, Chang CY, Nakamoto RK, Krugner-Higby L. Mitochondria in nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:595-617. [ Links ]
21. Postica C, Girarda J. The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes Metab. 2008;34:643-8. [ Links ]
22. Ginsberg HN, Zhang YL, Hernandez-Ono A. Regulation of plasma triglycerides in insulin resistance and diabetes. Arch Med Res. 2005;36:232-40. [ Links ]
23. Voshol PJ, Haemmerle G, Ouwens DM, Zimmermann R, Zechner R, Teusink B, et al. Increased hepatic insulin sensitivity together with decreased hepatic triglyceride stores in hormone-sensitive lipase-deficient mice. Endocrinology. 2003;144:3456-62. [ Links ]
24. Park SY, Kim HJ, Wang S, Higashimori T, Dong J, Kim YJ. Hormone-sensitive lipase knockout mice have increased hepatic insulin sensitivity and are protected from short-term diet-induced insulin resistance in skeletal muscle and heart. Am J Physiol Endocrinol Metab. 2005;289:30-9. [ Links ]
25. Utzschneider KM, Kahn SE. The role of insulin resistance in nonalcoholic fatty liver disease. J Clin Endocrinol Metab. 2006;91:4753-61. [ Links ]
26. Videla A, Rodrigo R, Araya J, Poniachik J. Insulin resistance and oxidative stress interdependency in non alcoholic fatty liver disease. Trends Mol Med. 2006;12:555-8. [ Links ]
27. Cave M, Deaciuc I, Mendez C, Song Z, Joshi-Barve S, Barve S, et al. Nonalcoholic fatty liver disease:predisposing factors and the role of nutrition. J Nutr Biochem. 2007;18:184-95. [ Links ]
28. Zulet A. Biomarcadores del estado inflamatorio, nexo de unión con la obesidad y complicaciones asociadas. Nutri Hosp. 2007;22:511-27. [ Links ]
29. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med. 2002;346:1221-31. [ Links ]
30. O'shea J, Gadina M, Schreiber R. Cytokine signaling in 2002: New surprises in the Jak/Stat pathway. Cell. 2002;109:S121-S31. [ Links ]
31. O'shea J, Murray P. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477-87. [ Links ]
32. Wullaert A, Van Loo G, Heyninck K, Beyaert R. Hepatic tumor necrosis factor signaling and nuclear factor kappa beta: effects on liver homeostasis and beyond. Endocrinol Rev. 2007;28:365-86. [ Links ]
33. Strasser H, McDunn A, Swanson J. Cell death. N Engl J Med. 2009;361:1570-83. [ Links ]
34. Aggarwal B. Signalling pathways of the TNF super family: a double-edged sword. Nat Rev Immunol. 2003;3:745-56. [ Links ]
35. Peraldi P, Hotamisligil. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55TNF receptor and activation of sphingomyelinase. J Biol Chem. 1996;271:13018-22. [ Links ]
36. Liu LM, Spelleken K, Rohring H, Hauner. Tumor necrosis factor-alpha acutely inhibits insulin signaling in human adipocytes:implication of the p80 tumor necrosis factor receptor. Diabetes. 1998;47:515-22. [ Links ]
37. Uysal K. Functional analysis of tumor necrosis factor (TNF) receptors in TNFalpha- mediated insulin resistance in genetic obesity. Endocrinology. 1998;139:4832-8. [ Links ]
38. Sethi J, Uysal S Wiesbrock L. Characterization of receptor-specific TNF-alpha functions in adipocyte cell lines lacking type 1 and 2 TNF receptors. FEBS Lett. 2000;469:77-82. [ Links ]
39. Hotamisligil GS, Arner P, Atkinson RL. Differential regulation of the p80 tumor necrosis factor receptor in human obesity and insulin resistance. Diabetes. 1997;46:451-5. [ Links ]
40. Hui JM, Hodge A, Farrell GC, Kench JG, Kriketos A, George J. Beyond insulin resistance in NASH:TNF-alpha or adiponectin? Hepatology. 2004;40:46-54. [ Links ]
41. Marra F, Aleffi S, Bertolani C, Petrai I, Vizzutti F. Adipokines and liver fibrosis. Eur Rev Med Pharmacol Sci. 2005;9:279-84. [ Links ]
42. Musso G, Gambino R, Biroli G, Carello M, Faga E, Pacini G. Hypoadiponectinemia predicts the severity of hepatic fibrosis and pancreatic Beta-cell dysfunction in nondiabetic nonobese patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2005;100:2438-46. [ Links ]
43. Trappoliere F, Loguercio C. Treatment of patients with non-alcoholic fatty liver disease: Current views and perspectives. Digest Liver Dis. 2006;38:789-80. [ Links ]
44. Lingvay I, Raskin P, Szczepaniak L. Effect of insulin–metformin combination on hepatic steatosis in patients with type 2 diabetes. J Diabetes Complications. 2007;21:137-42. [ Links ]
45. Capeau J. Insulin resistance and steatosis in humans. Diabetes Metab. 2008;34:649-57. [ Links ]
46. Bajaj M, Suraamornkul S, Piper P, Hardies LJ, Glass L, Cersosimo E. Decreased plasma adiponectin concentrations are closely related to hepatic fat content and hepatic insulin resistance in pioglitazone- treated type 2 diabetic patients. J Clin Endocrinol Metab. 2004;89:200-306. [ Links ]
47. Tiikkainen M, Hakkinen A, Korsheninnikova E, Nyman T, Makimattila S, Yki-Jarvinen H. Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes. 2004;53:2169-76. [ Links ]
48. Machado M, Cortez-Pinto H. Non-alcoholic fatty liver disease and insulin resistance. Eur J Gatroenterol Hepatol. 2005;17:823-6. [ Links ]
49. Ten S, Maclaren N. Insulin resistance syndrome in children. J Clin Endocrinol Metabol. 2004;89:2596-639. [ Links ]
50. Festi DA, Sacco T, Bondi M. Hepatic steatosis in obese patients: clinical aspects and prognostic significance. Obesity Rev. 2004;5:27-42. [ Links ]
51. Angulo P, Lindor K. Treatment of non-alcoholic steatohepatitis. Best Pract Res Clin Gastroenterol. 2002;16:797-810. [ Links ]
52. Hughes T. Emerging therapies for metabolic diseases: the focus is on diabetes and obesity. Curr Opin Chem Biol. 2009;13:1-6. [ Links ]

