










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkInvestigaciones Andina
Print version ISSN 0124-8146
Investig. andina vol.14 no.24 Pereira Apr. 2012
Artículo de Revisión
CARCINOGENESIS INDUCIDA POR EL VIRUS DEL PAPILOMA HUMANO
HUMAN PAPILLOMA VIRUS-INDUCED CARCINOGENESIS
CARCINOGÊNESE INDUZIDA PELO VÍRUS DO PAPILOMA HUMANO
Adalucy Álvarez Aldana, MSc*
Juan Carlos Sepúlveda Arias. MD., PhD*
Fernando Siller López, BSc., PhD**
* Doctorado en Ciencias Biomédicas, Facultad Ciencias de la Salud Universidad Tecnológica de Pereira.
** Docente, Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara.
Resumen
Introducción: la carcinogenesis es el proceso por el cual una célula normal se transforma en una célula cancerígena e involucra múltiples pasos, los cuales reflejan las alteraciones genéticas que conducen a la transformación progresiva del tejido normal hacia estados malignos. Los virus se han asociado con el desarrollo de cáncer, tanto en animales como en humanos, y dentro de estos se encuentra el Virus del Papiloma Humano (VPH), relacionado con el desarrollo del cáncer cervical, considerado el segundo tipo de cáncer en mujeres a nivel mundial. En esta revisión se describen los eventos responsables de la carcinogénesis inducida por el Virus del Papiloma Humano.
Métodos: se realizó una búsqueda sistemática de la literatura en la base de datos Medline.
Resultados: en esta revisión se discute la relación infección y cáncer, con énfasis en el proceso de carcinogénesis cervical inducido por el VPH y las moléculas involucradas en el mismo.
Conclusiones: las oncoproteínas E6 y E7 son primordiales en el proceso de transformación maligna inducida por el VPH e involucran muchos otros factores como la interacción de dichas proteínas con factores reguladores del ciclo celular.
Palabras clave: Cervix; Neoplasia; Papilomavirus Humano; Carcinogenesis.
Abstract
Introduction: carcinogenesis is the process by which a normal cell transforms into a cancer cell and involves multiple steps which reflect genetic alterations that lead to the progressive transformation of normal tissue to malignant states. Viruses have been associated with cancer development in both animals and humans. Human Papilloma viruses (HPV) are related with the development of cervical cancer, considered the second more prevalent type of cancer in women worldwide. In this review we describe the events responsible for the HPV-induced carcinogenesis.
Methods: a systematic literature search was conducted in Medline database.
Results: this review discusses the relationship between infection and cancer, with emphasis on the HPV-induced carcinogenesis and the molecules involved in this process.
Conclusions: E6 and E7 oncoproteins are essential in the process of malignant transformation induced by HPV and involve many other factors such as the interaction of these proteins with cell cycle regulatory factors.
Keywords: Cervix; Neoplasia; Human Papilloma virus; Carcinogenesis.
Resumo
Introdução: a carcinogênese é o processo, pelo qual, uma célula normal se transforma numa célula cancerígena e compreende vários passos, os quais refletem as alterações genéticas que conduzem à transformação progressiva do tecido normal até o estado de maligno. Os vírus associam-se ao desenvolvimento do câncer, tanto nos animais como nos humanos, e dentre deles encontra-se o Vírus do Papiloma Humano (VPH), relacionado com o desenvolvimento do câncer cervical, considerado o segundo tipo de câncer em mulheres no nível mundial. Nesta revisão descrevem-se os eventos responsáveis pela carcinogênese induzida pelo vírus do Papiloma Humano.
Métodos: realizou-se uma busca sistemática da literatura na base de dados Med-line.
Resultados: nesta revisão discute-se a relação infecção e câncer, com ênfase no processo de carcinogênese cervical induzido pelo VPH e as moléculas envolvidas no mesmo.
Conclusões: as oncoproteínas E6 e E7 são primordiais no processo de transformação maligna induzida pelo VPH e envolvem muitos outros fatores como a interação de tais proteínas com fatores reguladores do ciclo celular.
Palavras chave: Cérvix; Neoplasia; Papilomavírus Humano; Carcinogênese.
Fecha de recibo: Abril/2011
Fecha aprobación: Noviembre/2011
Introducción
Los papilomavirus humanos son un grupo de más de 70 tipos de virus causantes de las verrugas o papilomas; de allí derivan su nombre. Diferentes tipos de Virus del Papiloma Humano (VPH) causan las verrugas comunes que crecen en manos y pies, así como los que se desarrollan en boca y áreas genitales. Algunos VPH genitales, transmitidos sexualmente han sido asociados a procesos neoplásicos tanto en mujeres como en hombres. Existe una relación causal entre infecciones persistentes con genotipos de alto riesgo (tipos 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66) y el desarrollo de cáncer cervical.
Los virus son parásitos intracelulares obligados que necesitan de una célula huésped para completar su ciclo de vida, y dentro de esta relación, las células han desarrollado estrategias para controlar la replicación viral, y a su vez los virus han desarrollado mecanismos para evadir la respuesta antiviral de la célula infectada. De esta manera los virus aprendieron a sobrevivir en un ambiente celular hostil.
Se calcula que aproximadamente un 18% de los cánceres humanos tienen etiología viral. Dentro de estos, el cáncer cervical ha sido ampliamente estudiado y el factor de riesgo mejor caracterizado para el desarrollo de dicho cáncer es la integración del ADN del VPH en los cromosomas de las células del cuello uterino. En las células infectadas, las proteínas virales alteran la actividad de las proteínas de dicha célula, disregulando el ciclo celular e induciendo los cambios neoplásicos.
La presente revisión pretende compilar la información actual con relación al proceso de carcinogénesis con énfasis en los cambios inducidos por la infección con el VPH.
Materiales y métodos
Se realizó una búsqueda sistemática de la literatura en la base de datos Medline, utilizando los términos Cervical carcinogenesis y Human papillomavirus, para lo cual se empleó el conector Booleano AND. La búsqueda arrojó como resultado 1040 artículos, de los cuales 852 fueron trabajos originales (321 de acceso libre) y 188 fueron revisiones. Se restringió la bibliografía a trabajos originales publicados principalmente en los últimos 12 años, revisiones y unos pocos artículos originales clásicos con relación altemapublicadocon anterioridad a 1999. Se seleccionaron artículos que evaluaran el proceso de carcinogénesis en el cuello uterino, inducido por el VPH. Se incluyeron algunas referencias generales con relación al proceso de carcinogénesis y a la relación infección-cáncer.
Resultados
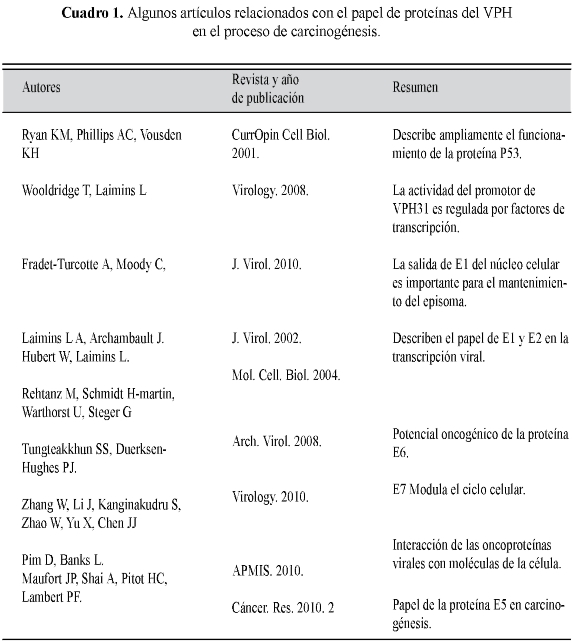
En el cuadro 1 se describen algunos de los artículos consultados y una corta descripción de los hallazgos.
Discusión
Antecedentes históricos
El concepto de crecimiento canceroso es muy antiguo y se han postulado diferentes teorías para explicar el desarrollo del cáncer, pero fue solo hasta el siglo XX que se consideró la teoría sobre la etiología infecciosa del mismo (1).
A pesar de la evidencia que sugería cómo agentes infecciosos sub-microscópicos estaban asociados con cáncer, la aceptación de esta teoría tomó muchos años. M'Fadyan y Hobday describieron en 1898 la transmisión de verrugas orales de perro con extractos libres de células, y Ciuffo publicó estudios de transmisión similares con verrugas humanas en 1907. El significado de estos hallazgos no fue apreciado ya que las verrugas son hiperplasias benignas y no tumores malignos. En 1908, Ellermann y Bang demostraron que la leucemia en pájaros puede ser transmitida de animal a animal vía extractos de células leucémicas o de suero de aves enfermas. Sin embargo, en el momento no se dieron cuenta de que se trataba de la primera transmisión con éxito de un tumor de origen natural, ya que la leucemia no había sido aceptada todavía como cáncer (2).
En 1911, Peyton Rous produce tumores sólidos en pollos usando extractos libres de células a partir de un sarcoma (3). Este estudio también fue visto con bastante escepticismo debido a que no era considerado un modelo válido para cáncer humano. De hecho, la importancia de este estudio no se apreció del todo hasta la conclusión de que las leucemias de ratón pueden ser inducidas por virus. En las siguientes dos décadas numerosos virus de tumores animales fueron aislados, y en 1966 Rous fue reconocido con el premio Nobel por su trabajo pionero sobre virus tumorales animales (2).
El análisis de la relación de causalidad entre infección viral y leucemia en ratones (Gross, 1950), la demostración posterior de los retrovirus como los factores causales y la presencia de virus similares, particularmente en trastornos linfoproliferativos en bovinos, gatos, y varias especies de roedores, justificó durante décadas la búsqueda de agentes similares en los cánceres humanos (4). De este modo, en 1964, el virus de Epstein-Barr(VEB)fueobservadopormicroscopia electrónica en células cultivadas de linfoma de Burkitt y en 1970 el virus de hepatitis B (VHB) fue visualizado en suero humano positivo para antígeno Australia (designado actualmente como antígeno de superficie del VHB). Posteriormente se detectaron diversos posibles virus tumorales humanos que incluyen al virus linfotrópico de células T (HTLV-I), el virus de la hepatitis C (VHC) y el virus herpes humano 8 (VHH 8) también conocido como virus herpes asociado al sarcoma de Kaposi (VHKS) (5). El panorama actual es de un mayor número de agentes infecciosos asociados al desarrollo de cáncer (6).
Carcinogénesis
La carcinogénesis es el proceso por el cual una célula normal se transforma hacia una célula cancerígena. La carcinogénesis puede ser vista como un proceso que involucra mutaciones secuenciales que le confiere a las células mutadas dominancia en el crecimiento sobre las células vecinas normales, resultando en un incremento de las células mutadas. La carcinogénesis es un proceso de múltiples pasos y estos pasos reflejan las alteraciones genéticas que conducen a la transformación progresiva del tejido normal hacia estados malignos (7). En el cáncer, la regulación del crecimiento y la madurez de las células están alteradas. Existen numerosos tipos de cáncer e incluso el cáncer del mismo tipo se puede comportar de manera muy diferente en cada individuo. A pesar de estas diferencias, hay cambios fundamentales comunes a todos los prototipos de cáncer como lo menciona Harold Varmus, "las células cancerosas se dividen sin restricción, cruzan las fronteras que estaban destinadas a respetar, y no muestran las características del linaje de las células de las que se derivan" (8).
En el cáncer, los cambios citogenéticos progresan a través de una serie de mutaciones somáticas secuenciales en genes específicos. Esto puede ser causado por exposición a uno o más de una variedad de agentes químicos o físicos, errores en la replicación génica, alteraciones en los genes que controlan la progresión del ciclo celular y los procesos de reparación del ADN. Las alteraciones somáticas junto con los eventos epigenéticos que se refieren a cambios en la expresión (fenotipo) sin presentar alteraciones en la estructura del ADN (genotipo), determinan el desarrollo de la transformación maligna (9).
La proliferación celular se asocia al evento denominado ciclo de división celular o ciclo celular (CC), proceso por el cual una célula crece, replica su ADN y se divide para dar origen a dos células hijas. Este proceso se clasifica en cuatro fases secuenciales denominadas G1, G2, S y M. S es la fase de síntesis del ADN y M es la fase de división celular. Existe una quinta fase conocida como G0 o de quiescencia en la cual la célula se mantiene reversiblemente por fuera del CC (Figura 1). Existen puntos de control en los cuales la célula monitorea el ambiente celular y determina si hay condiciones apropiadas para progresar a través del ciclo celular. Los puntos de control son clave para el funcionamiento correcto del CC y los fallos en su funcionamiento pueden dar lugar a que las células se dividan sin control (10).
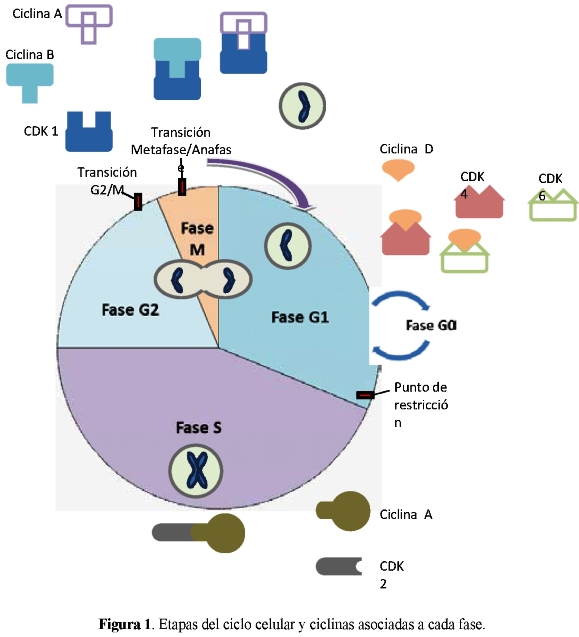
El control del ciclo celular es llevado a cabo por componentes fundamentales como las CDKs o cinasas dependientes de ciclina (cdk2, cdk4, cdk6, cdc2/cdk1), las ciclinas (ciclinas A, B, D y E), inhibidores de CDKs, junto con proteínas supresoras de tumores como p53 y la familia pRB. Cada una de las ciclinas forma complejos con su respectiva CDKs, activándoles la actividad cinasa. La proteína Rb en la forma hipofosforilada bloquea a los factores de transcripción E2F1, E2F2 y E2F3a, esenciales para la expresión de genes que le dan continuidad al ciclo celular, mientras que la fosforilación parcial de Rb deja en libertad a estos factores de transcripción (11). La proteína p53 actúa en el CC evitando que la célula entre a fase S ante daño del ADN, deteniendo el ciclo en G1. Si el daño es producido luego de la replicación del ADN (fase S), la célula se detiene en la fase G2. Cuando el daño del ADN es irreparable, p53 lleva a la muerte celular por apoptosis. Estas proteínas supresoras de tumores permiten un control preciso del ciclo celular y bloquean eventos carcinogénicos (12).
Infección y cáncer
Se reporta que el 17,8% (1.9 millones de casos) de todos los casos de cáncer son debidos a agentes infecciosos, que incluyen virus (12.1%), bacterias (5.6%) y helmintos (0.1%), siendo esta frecuencia mayor en países en vía de desarrollo (26,3%) (1.5 millones de casos) (6,13). Se han descrito como agentes biológicos carcinógenos siete virus (mencionados más adelante), una bacteria (Helicobacter pylori) y tres helmintos (Schistosoma haematobium, Opistorchis viverrini, y Clonorchis sinensis) (6). Es importante mencionar que dichos helmintos no se encuentran en América Latina.
Los principales mecanismos por los cuales la infección crónica promueve el cáncer se debe a la interacción que ocurre entre el huésped y el patógeno (14). Los agentes infecciosos pueden promover y mantener la formación de un tumor por mecanismos que pueden ser divididos en tres categorías; directa, indirecta por inflamación crónica o indirecta por supresión inmune crónica (6, 15).
La categoría directa se da en aquellos agentes infecciosos que inducen transformación como algunos virus, ya sea por la persistencia del genoma viral sin replicación de forma latente como el caso del VEB, que infecta los linfocitos B, o a través de la integración del genoma viral al genoma de la célula hospedera como es el caso del VPH que causa cáncer de cuello uterino. Este proceso de transformación ocurre por la presencia de proteínas virales que promueven el crecimiento celular y la supervivencia de la célula al intervenir en el ciclo celular. La categoría indirecta por inflamación crónica ocurre como resultado de una continua respuesta inmune debido a una infección persistente, como ocurre con los virus de la hepatitis B y C, los helmintos S. haematobium, O. viverrini, C. sinensis y la bacteria Gram negativa Helicobacter pylori. La prevalencia de la infección por H. pylori a nivel mundial es del 75%, sin embargo, no todas las personas infectadas con H. pylori desarrollan cáncer gástrico por lo que el agente infeccioso es un factor de riesgo indicando que factores de tipo genético y ambiental están involucrados en el desarrollo de este tipo de cáncer. El mecanismo indirecto por supresión crónica del sistema inmune ocurre en el caso del VIH. Un sistema inmune comprometido puede resultar en un incremento en la incidencia de tumores, como ha sido observado en individuos VIH positivos o en pacientes trasplantados que son tratados con inmunosupresores (15, 16, 17).
Los virus son agentes etiológicos importantes en cáncer humano, ya que del 17,8% de todos los casos de cáncer debidos a agentes infecciosos, el 12,1% corresponde a agentes virales (13). Hay siete virus implicados en cáncer humano: cuatro de estos actúan como carcinógenos directos como el HTLV-I, VPH, VEB y el VHH8/VHKS. Dos actúan como carcinógenos indirectos vía inflamación crónica, el VHB y el VHC. Uno, el VIH-1, actúa como carcinógeno indirecto vía inmunosupresión crónica (6, 18, 19). El HTLV-I es un retrovirus que causa leucemia de células T (20); el VPH induce cáncer de cuello uterino (21,22); VHH 8 es el agente causal del sarcoma de Kaposi (23); el VHB y el VHC inducen carcinoma hepatocelular, mientras que el VEB se asocia entre otros con linfoma de Burkitt y carcinoma nasofaríngeo (19, 24, 25).
El papel del VPH en procesos carcinogénicos de la región genital ha sido extensivamente investigado y documentado y es el propósito de esta revisión.
El virus del papiloma humano induce carcinogénesis
El VPH infecta células epiteliales basales, causando lesiones benignas y malignas de la piel y las mucosas. Se han identificado 150 genotipos, de los cuales 40 son considerados de tipo genital. Entre estos los genotipos VPH-16, -18, -31, -33, -35, -39, -45, -51, -52, - 56, -58, -59, y -66 son considerados de alto riesgo debido a su relación causal con cáncer cervical y otros tipos de cáncer anogenital (6, 26, 27, 28).
El genoma del VPH consiste de una molécula de ADN circular de doble cadena, con un rango de tamaño entre 7600 - 8000 pb, que se replica en el núcleo de las células infectadas como un plásmido de múltiples copias. El genoma está dividido en cuatro regiones (Figura 2). La región larga de control (LCR) o de regulación corriente arriba (URR), que no contiene marco de lectura alguno, donde se ubican el origen de replicación y elementos cis de la regulación como promotores y potenciadores; la región que corresponde a las proteínas de expresión temprana (E1-E2 y E4-E7) relacionadas con la replicación (E1) transcripción (E2) y transformación celular (E6 y E7), que se expresan en células transformadas basales de las lesiones; la región que corresponde a las proteínas de expresión tardía (L1 y L2) que constituyen la cápside viral y se expresan únicamente en los queratinocitos diferenciados de la capa superficial de la lesión, donde se producen partículas víricas maduras; y una región pequeña, altamente variable y no codificante (NCR) entre los genes L5 y E2 (22, 26, 29).
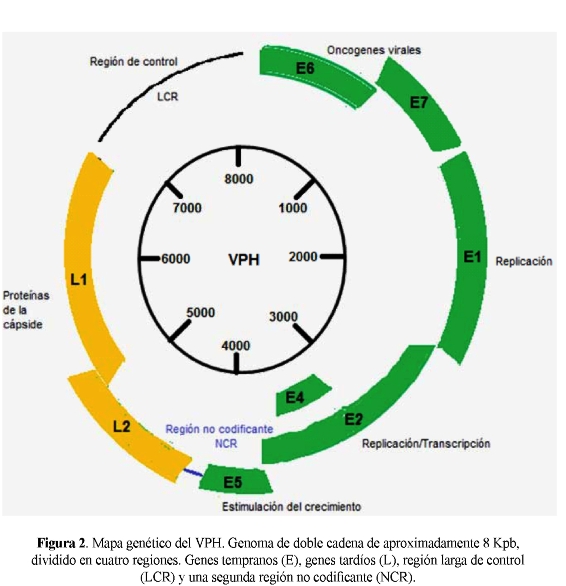
La cápside viral es de simetría icosahédrica sin envoltura y está formada por dos proteínas: la proteína L1 o mayor, de 55 kDa y que representa el 80% de la cápside, y la proteína L2 o menor, de 74 kDa. El diámetro de la cápside es de aproximadamente 55 a 60 nm y está constituida por 72 capsómeros pentaméricos, cada uno constituido por 5 monómeros de la proteína L1. De los 72 capsómeros, 60 interactúan en simetría seis, mientras que los 12 restantes lo hacen en simetría cinco (30, 31).
La expresión está regulada por la diferenciación celular y tiene lugar en la misma dirección 5'D3' usando múltiples promotores. Todos los VPH tienen dos promotores principales: P1 que está situado inmediatamente corriente arriba del gen E6 y P3 localizado dentro del gen E7 (32). La región E (temprana) y L (tardía) son seguidas de sitios de poliadenilación (Figura 3). El promotor temprano controla la expresión en células no diferenciadas y dirige la iniciación de la transcripción viral en regiones localizadas corriente arriba del marco de lectura abierto (ORF) para E6. La expresión a partir de este promotor genera transcritos policistrónicos, que codifican las proteínas E6, E7, E1, E2, E4 y E5, las cuales terminan en regiones de poliadenilación localizadas corriente abajo del ORF para E5. Este promotor se denomina P97 en VPH31 y es análogo al promotor P97 de VPH16 y P105 de VPH18. Se regula mediante factores de transcripción que se unen a secuencias de inicio de la región reguladora (LCR) (33,34).
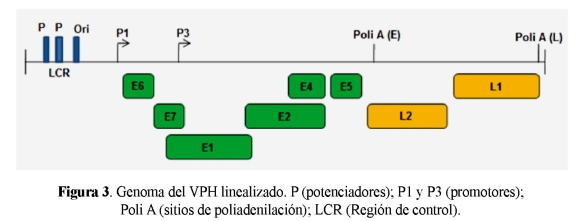
El promotor tardío, denominado P742, dirige la expresión de los transcritos que codifican las proteínas tardías, incluyendo E1, E4, E5, L1 y L2. Estos transcritos se activan durante la diferenciación, iniciándose en diferentes regiones localizadas alrededor del nucleótido 742 en VPH31 (33). Se han identificado secuencias en la región E6/E7 así como en la región LCR que regulan este promotor tardío (35, 36). También se han identificado varios promotores secundarios en las regiones E6/E7 que son activados durante la diferenciación (37).
El ciclo de vida del VPH se acopla con el programa de diferenciación de los queratinocitos en el epitelio. Los pseudoviriones inicialmente se unen a la capa basal cervico-vaginal en los sitios de trauma donde la cápside sufre un cambio conformacional por la acción de una furina que cliva la proteína L2. De este modo el virus se une a los receptores de membrana tipo integrinas de las células de la lámina basal (38). Después de la infección, el virus establece y mantiene su genoma como un elemento extracromosomal (episoma) en el núcleo de las células infectadas. El episoma es mantenido en bajo número de copias pero este se incrementa en las capas superiores del epitelio por la acción de los oncogenes virales E6 y E7 (39).
Las células de la capa basal son células madre que están constantemente dividiéndose para dar origen a las células de la región suprabasal (40). La infección por el VPH de estas células lleva a la activación de la expresión de una cascada de genes virales que resulta en la producción de aproximadamente 20 a 100 copias extracromosómicas del virus por célula infectada. Entre las primeras proteínas virales expresadas se encuentran los factores de replicación E1 y E2, los cuales forman un complejo, uniéndose al origen de replicación viral y actúan reclutando polimerasas y proteínas accesorias que median replicación en la célula (41,42). La proteína E1 también exhibe actividad helicasa permitiendo la separación de las cadenas del ADN viral en el complejo de replicación (43). La proteína E2 es una proteína de unión al ADN que ayuda a reclutar a E1 al origen de replicación, pero también juega un papel en la regulación de la transcripción viral (44).
Las proteínas E6 y E7 de los VPH de alto riesgo actúan como oncoproteínas virales, por lo que tienen un papel fundamental en la carcinogénesis mediada por el VPH. La primera indicación del papel en carcinogénesis de estas proteínas surgió de las líneas celulares derivadas de cáncer cervical SiHa y CaSki, en las cuales,el ADN viral está integrado aleatoriamente en el genoma de la célula. Además, la integración del ADN viral resulta en la pérdida o alteración de algunos genes virales, excepto los genes E6 y E7, los cuales se transcriben activamente (45). E6 y E7 interfieren activamente en el ciclo celular, apoptosis y mantenimiento de la estabilidad cromosomal al interactuar con las proteínas supresoras de tumores p53 y pRB, respectivamente.
La proteína E6 del VPH codifica para una proteína de 151 aminoácidos con un peso molecular de 16 a 18 kDa. E6 es una de las proteínas que se expresan tempranamente durante una infección por VPH. Esto le confiere varias funciones que alteran el ambiente celular, como por ejemplo: el bloqueo de la apoptosis mediante la degradación de p53, la alteración de la transcripción de genes celulares a través de la interacción con p300 y CBP y el incremento de la vida celular por la sobre activación de la telomerasa. La acción clave de E6 en los VPH de alto riesgo es inhibir la función de p53, mediante su degradación por la vía de la ubiquitina con la participación de la proteína celular asociada a E6 (E6-AP) (Figura 4) (4549).
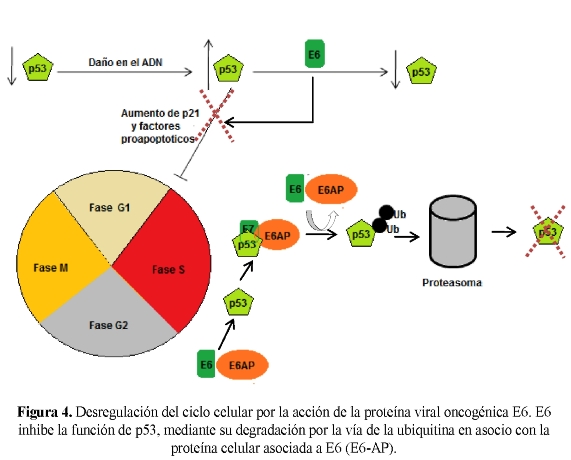
La proteína p53 se activa cuando el ADN celular sufre algún daño, uniéndose a él e induciendo así la expresión del gen que codifica la proteína p21. Dicha proteína es un inhibidor de ciclinas dependientes de cinasas (CDK), que se une a los complejos de ciclina G1 con proteína CDK2, la cual conduce a la célula más allá del punto de control G1 del ciclo celular. La proteína p21 bloquea la actividad cinasa de estos complejos, evitando así la progresión de la célula en fase S y la replicación de su ADN. Las células no podrán progresar en la fase S del ciclo celular y se retrasarán en la fase G1 o morirán por apoptosis (11). Por tanto, la degradación de p53 tiene como consecuencia la evasión del punto de control del ciclo celular G1/S, dando lugar a la desregulación del ciclo celular.
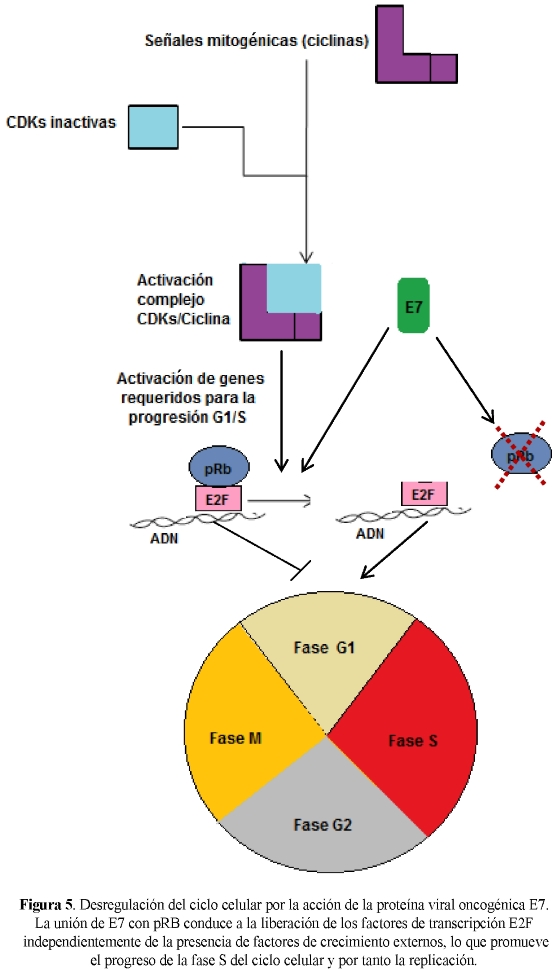
La proteína E7 del VPH codifica para una proteína de aproximadamente 98 aminoácidos con un peso molecular de 10 kDa (45). E7 tiene la mayor capacidad transformante y actúa mediante la unión a proteínas celulares supresoras de tumores de la familia pRB, que a su vez interactúan con factores de transcripción de la familia E2F (50). La familia pRB controla la replicación celular (11). La unión de E7 con pRB conduce a la liberación de los factores de transcripción E2F independientemente de la presencia de factores de crecimiento externos, lo que promueve el progreso de la fase S del ciclo celular y por tanto la replicación celular (Figura 5). E7 también se asocia con otras proteínas tales como desacetilasas de histonas, AP1 e inhibidores de los complejos CDK, como p21 y p27. Como resultado de la liberación de E2F se expresa ciclina E, importante para el progreso de la fase S. Estas interacciones inducen múltiples respuestas celulares, incluyendo la disminución de p53 que normalmente contrarrestaría esta replicación celular, anormalmente estimulada, mediante el incremento de la apoptosis. Sin embargo, la proteína E6 degrada a p53 y por tanto bloquea esta respuesta celular (50-52).
Con la división de las células basales infectadas por el VPH, los genomas virales son distribuidos en las células hijas, estas células se separan de la capa basal, migran hacia el estrato granuloso y sufren diferenciación. Cuando las células epiteliales normales no infectadas migran hacia las capas suprabasales, salen del ciclo de división celular. Sin embargo, cuando las células infectadas migran hacia las capas suprabasales, se mantienen activas en el ciclo celular debido a la acción de la proteína E7, corroborando su papel transformante en las células infectadas (53). Estas células entran en fase S y activan la expresión de factores de replicación celular requeridos para la replicación viral. Las oncoproteínas virales E6 y E7 no solo son necesarias para la inmortalización y retención de las células en el ciclo celular, sino también son necesarias para el mantenimiento extracromosomal del VPH en células basales no diferenciadas (54).
En lesiones de bajo grado, los genomas de VPH de alto riesgo están presentes en episomas mientras progresan las lesiones hacia alto grado o carcinomas, donde el genoma viral es integrado en el genoma del hospedero. Esta integración usualmente ocurre dentro del gen E2 dando como resultado la pérdida del control de la transcripción que ejerce la proteína E2, incrementando los niveles de expresión de E6 y E7 (55, 56).
Las oncoproteínas E6 y E7 han sido ampliamente estudiadas y son importantes en carcinogénesis anogenital por la desregulación del ciclo celular (57,58), pero poco se conoce acerca de la proteína E5. Datos recientes indican que E5 actúa como un oncogén en el tracto reproductivo de ratones hembra (59). E5 es una proteína de membrana hidrofóbica que se halla principalmente en el retículo endoplásmico y aparato de Golgi, pero también en la membrana citoplasmática y nuclear (60,61). "itemData": {"abstract": "The E5 oncoprotein encoded by bovine papillomavirus type 1 is a homodimeric, hydrophobic polypeptide which is localized predominantly in Golgi membranes and which transforms several cell types apparently by inducing tyrosine phosphorylation of the platelet-derived growth factor receptor (PDGF-RE5 regula la actividad de los receptores de factores de crecimiento, como el del factor de crecimiento epidérmico (EGFR) o el de crecimiento derivado de las plaquetas (PDGFR) (62). E5 se une a la subunidad de 16 KDa de la ATPasa vacuolar inhibiendo la maduración de los endosomas (63), lo que podría también afectar la presentación antigénica y así disminuir el reconocimiento inmune del virus (64,65). Los queratinocitos que expresan E5 incrementan la producción de EGFR, explicando el papel de E5 en proliferación celular (66). En células de roedores que expresan E5 se presenta un aumento en la expresión de c-Fos y c-Jun, en especial en presencia de EGF (67).
La proteína E4, se expresa desde las fases tempranas de la infección pero de manera abundante durante las etapas tardías del ciclo viral (68). La expresión es previa a la síntesis de las proteínas de la cápside viral y el ensamblaje de las partículas virales. La proteína E4 se localiza en los filamentos intermedios de la queratina del citoplasma, y de manera difusa en regiones perinuclares y citoplasmáticas. E4 causa el colapso de las queratinas y esto se ha relacionado con la liberación de los viriones (69). E4 induce paro en la fase G2 del ciclo celular cuando se expresa en células HeLa y SiHa. Esto sugiere un papel antagónico con la proliferación celular inducida por E7 durante las etapas tempranas de la infección (70). E4 incrementa o disminuye la transcripción mediada por E2, dependiendo de las concentraciones relativas de ambas proteínas. Esta evidencia sugiere que durante la fase infecciosa productiva de viriones, E4 regula los niveles de la proteína E2 para facilitar la amplificación del genoma viral y la expresión de proteínas tempranas (71).
Actualmente, es difícil asignar un papel para las proteínas tempranas como E1, E2 y E4 en el proceso de carcinogénesis mediada por la infección con el VPH.
Los estudios epidemiológicos y experimentales de la biología del VPH sobre la relación entre cáncer y VPH, muestran una historia aun sin concluir. Pero, tener cada vez más información sobre el papel de cada una de las proteínas del virus a nivel del ciclo viral, en el ciclo de la célula huésped y en su propia regulación de la expresión génica, nos va mostrando un horizonte que quizás nos permita en el futuro, detener los eventos carcinogénicos en los que el VPH está implicado.
Agradecimientos
Los autores agradecen a COLCIENCIAS por el apoyo financiero de la estudiante de Doctorado y del proyecto de investigación (contrato 371-2008) en el marco del cual se realizó esta revisión. A la Universidad Tecnológica de Pereira por el apoyo financiero al Dr. Fernando Siller-López para su estancia en el Grupo Infección e Inmunidad.
REFERENCIAS
1. Ronald TJ, Butel JS. The History of Tumor Virology. Cancer Research. 2008; 68: 7693-7706. [ Links ]
2. McLaughlinDrubbin M, Munger K. Viruses Associated with Human Cancer. Biochim Biophys Acta. 2008; 1782(3):127-150. [ Links ]
3. Becsei-Kilborn E. Scientific Discovery and Scientific Reputation: The Reception of Peyton Rous' Discovery of the Chicken Sarcoma Virus. Journal of the History of Biology. 2010; 43(1):111-157. [ Links ]
4. Hausen H zur. Oncogenic DNA viruses. Oncogene. 2001; 20:7820-3. [ Links ]
5. Butel JS. Viral Carcinogénesis: Revelation of Molecular Mechanisms and etiology of human disease. Carcinogénesis 2000; 21(3):405-26. [ Links ]
6. IARC Monographs: A Review of Human Carcinogens, Part B Biological Agents. In IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. 2009. Volume 100. IARC, Lyon,France. [ Links ]
7. Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100:57-70 [ Links ]
8. Peters J, Loud J, Dimond E, Jenkins J. Cancer genetics fundamentals. Cancer Nursing. 2001;24(6):446-61. [ Links ]
9. Feller L, Wood NH, Khammissa RAG, Lemmer J. Human papillomavirus-mediated carcinogénesis and HPV-associated oral and oropharyngeal squamous cell carcinoma. Part 1: human papillomavirus-mediated carcinogénesis. Head & Face Med. 2010; 6:14. [ Links ]
10. Garret MD. Cell cycle control and cancer. Current Science. 2001; 81(5):515-22. [ Links ]
11. Quezada MA. El ciclo celular, sus alteraciones en el cáncer y como es regulado en células troncales embrionarias. ContactoS. 2007; 65:5-12. [ Links ]
12. Ryan KM, Phillips AC, Vousden KH. Regulation and function of the p53 tumor suppressor protein. Curr Opin Cell Biol. 2001; 13(3):332-337. [ Links ]
13. Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006; 118(12):3030-44. [ Links ]
14. Ou HD, May AP, O'Shea CC. The critical protein interactions and structures that elicit growth deregulation in cancer and viral replication. Interdiscip Rev Syst Biol Med. 2011; 3(1):48-73. [ Links ]
15. Dalton-Griffin L, Kellam P. Infectious causes of cancer and their detection. J Biol. 2009; 8(7):67. [ Links ]
16. Lax AJ, Thomas W. How bacteria could cause cancer: one step at a time. Trends Microbiol. 2002;10(6):293-9. [ Links ]
17. Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet. 2007; 370: 59-67. [ Links ]
18. Bergonzini V, Salata C, Calistri A, Parolin C, Palú G. View and review on viral oncology research. Infect Agent Cancer. 2010; 5:11. [ Links ]
19. Yasunaga J, Jeang K-T. Viral transformation and aneuploidy. Environ Mol Mutagen. 2009; 50(8):733-740. [ Links ]
20. Matsuoka M, Jeang K-T. Human T-cell leukemia virus type 1 (HTLV-1) and leukemic transformation: viral infectivity, Tax, HBZ and therapy. Oncogene. 2011; 30(12):1379-89. [ Links ]
21. Woodman CBJ, Collins SI, Young LS. The natural history of cervical HPV infection: unresolved issues. Nat Rev Cancer. 2007; 7(1):11-22. [ Links ]
22. Muñoz N, Castellsagué X, de González AB, Gissmann L. Chapter 1: HPV in the etiology of human cancer. Vaccine. 2006; 24 Suppl 3:S3/1-10. [ Links ]
23. Cathomas G. Human herpes virus 8: a new virus discloses its face. Virchows Arch. 2000;436(3):195-206. [ Links ]
24. Carrillo-Infante C, Abbadessa G, Bagella L, Giordano A. Viral infections as a cause of cancer. Int JOncol. 2007; 30(6):1521-8. [ Links ]
25. Chang MH. Hepatitis B virus and cancer prevention. Recent Results Cancer Res. 2011; 188: 75-84. [ Links ]
26. zur Hausen H. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogénesis. J Natl Cancer Inst. 2000; 92(9):690-8. [ Links ]
27. Muñoz N, Bosch FX, de Sanjosé S, Herrero R, Castellsagué X, Shah KV et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003; 348(6):518-27. [ Links ]
28. Schiffman 2009 Schiffman M, Clifford G, Buonaguro FM. Classification of weakly carcinogenic human papillomavirus types: addressing the limits of epidemiology at the borderline. Infect Agent Cancer. 2009. 1; 4:8. [ Links ]
29. Smith 2011 Smith B, Chen Z, Reimers L, van Doorslaer K, Schiffman M, Desalle R, et al. Sequence imputation of HPV16 genomes for genetic association studies. PLoS One. 2011; 6(6):e21375. [ Links ]
30. Vanegas VA, Rubio AI, Bedoya AM, Sanchez G. Antigenic and Molecular Structure of Human Papillomavirus (HPV) 16 Vaccine. Acta Biol Colomb. 2008; 13(3):37-48. [ Links ]
31. Modis Y, Trus BL, Harrison SC. Atomic model of the papillomavirus capsid. Embo J. 2002; 21(18):4754-62. [ Links ]
32. Chow L, Broker T, Steinberg B. The natural history of human papillomavirus infections of the mucosal epithelia. APMIS. 2010; 118(6-7):422-49. [ Links ]
33. Wooldridge T, Laimins L. Regulation of human papillomavirus type 31 gene expression during the differentiation-dependent life cycle through histone modifications and transcription factor binding. Virology. 2008; 374(2):371-80. [ Links ]
34. Demeret C, Yaniv M, Thierry F. The E2 transcriptional repressor can compensate for Sp1 activation of the human papillomavirus type 18 early promoter. J Virol. 1994; 68(11):7075-82. [ Links ]
35. Bodily JM, Meyers C. Genetic Analysis of the Human Papillomavirus Type 31 Differentiation-Dependent Late Promoter. J Virol. 2005; 79(6):3309-3321. [ Links ]
36. Spink KM, Laimins LA. Induction of the Human Papillomavirus Type 31 Late Promoter Requires Differentiation but Not DNA Amplification. J Virol. 2005; 79(8):4918-4926. [ Links ]
37. Ozbun M, Meyers C. Two novel promoters in the upstream regulatory region of human papillomavirus type 31b are negatively regulated by epithelial differentiation. J Virol. 1999; 73(4):3505-10. [ Links ]
38. Kines RC, Thompson CD, Lowy DR, Schiller JT, Day PM. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. PNAS. 2009; 106(48):20458-63. [ Links ]
39. Fradet-Turcotte A, Moody C, Laimins L a, Archambault J. Nuclear export of human papillomavirus type 31 E1 is regulated by Cdk2 phosphorylation and required for viral genome maintenance. J Virol. 2010; 84(22):11747-60. [ Links ]
40. Hummel M, Hudson JB, Laimins L a. Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J Virol. 1992; 66(10):6070-80. [ Links ]
41. Hubert W, Laimins L. Human Papillomavirus Type 31 Replication Modes during the Early Phases of the Viral Life Cycle Depend on Transcriptional and Posttranscriptional Regulation of E1 and E2 Expression. J Virol. 2002; 76(5):2263-2273. [ Links ]
42. Rehtanz M, Schmidt H-martin, Warthorst U, Steger G. Direct Interaction between Nucleosome Assembly Protein 1 and the Papillomavirus E2 Proteins Involved in Activation of Transcription. Mol Cell Biol. 2004; 24(5):2153-2168. [ Links ]
43. Chen G, Stenlund A. Sequential and Ordered Assembly of E1 Initiator Complexes on the Papillomavirus Origin of DNA Replication Generates Progressive Structural Changes Related to Melting. Mol Cell Biol. 2002; 22(21):7712-7720. [ Links ]
44. Yu T, Peng Y-C, Androphy EJ. Mitotic kinesin-like protein 2 binds and colocalizes with papillomavirus E2 during mitosis. J Virol. 2007; 81(4):1736-45. [ Links ]
45. Ghittoni R, Accardi R, Hasan U, Gheit T, Sylla B, Tommasino M. The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes. 2010; 40(1):1-13. [ Links ]
46. Tommasino M, Accardi R, Caldeira S, Dong W, Malanchi I, Smet A, et al. The role of TP53 in Cervical carcinogénesis. Hum Mutat. 2003; 21(3):307-12. [ Links ]
47. Sekaric P, Cherry JJ, Androphy EJ. Binding of human papillomavirus type 16 E6 to E6AP is not required for activation of hTERT. J Virol. 2008; 82(1):71-6. [ Links ]
48. Tungteakkhun SS, Duerksen-Hughes PJ. Cellular binding partners of the human papillomavirus E6 protein. Arch Virol. 2008; 153(3):397-408. [ Links ]
49. Tungteakkhun SS, Filippova M, Fodor N, Duerksen-Hughes PJ. The full-length isoform of human papillomavirus 16 E6 and its splice variant E6* bind to different sites on the procaspase 8 death effector domain. J Virol. 2010; 84(3):1453-63. [ Links ]
50. Zhang W, Li J, Kanginakudru S, Zhao W, Yu X, Chen JJ. The human papillomavirus type 58 E7 oncoprotein modulates cell cycle regulatory proteins and abrogates cell cycle checkpoints. Virology. 2010; 397(1):1-14. [ Links ]
51. Flores ER, Allen-Hoffmann BL, Lee D, Lambert PF. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J Virol. 2000; 74(14):6622-31. [ Links ]
52. Spardy N, Covella K, Cha E, Hoskins EE, Wells SI, Duensing A, et al. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009; 69(17):7022-9. [ Links ]
53. Cheng S, Schmidt-Grimminger DC, Murant T, Broker TR, Chow LT. Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev. 1995; 9(19):2335-2349. [ Links ]
54. Thomas J, Hubert WG, Ruesch MN, Laimins L a. Human papillomavirus type 31 oncoproteins E6 and E7 are required for the maintenance of episomes during the viral life cycle in normal human keratinocytes. PNAS. 1999; 96(15):8449-54. [ Links ]
55. Jeon S, Allen-Hoffmann BL, Lambert PF. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J Virol. 1995; 69(5):2989-97. [ Links ]
56. Schneider-Gadicke A, Schwarz E. Different human cervical carcinoma cell lines show similar transcription patterns of human papillomavirus type 18 early genes. EMBO J. 1986; 5(9):2285-92. [ Links ]
57. Pim D, Banks L. Interaction of viral oncoproteins with cellular target molecules: infection with high-risk vs low-risk human papillomaviruses. APMIS. 2010; 118(6-7):471-93. [ Links ]
58. Kim YT, Zhao M. Aberrant cell cycle regulation in cervical carcinoma. Yonsei Med J. 2005; 46(5):597-613. [ Links ]
59. Maufort JP, Shai A, Pitot HC, Lambert PF. A role for HPV16 E5 in cervical carcinogénesis. Cancer Res. 2010; 70(7):2924-31. [ Links ]
60. Tsai T-C, Chen S-L. The biochemical and biological functions of human papillomavirus type 16 E5 protein. Arch Virol. 2003;148(8):1445-53. [ Links ]
61. Sparkowski J, Anders J, Schlegel R. E5 oncoprotein retained in the endoplasmic reticulum/cis Golgi still induces PDGF receptor autophosphorylation but does not transform cells. EMBO J. 1995; 14(13):3055-63. [ Links ]
62. DiMaio D, Mattoon D. Mechanisms of cell transformation by papillomavirus E5 proteins. Oncogene. 2001; 20(54):7866-73. [ Links ]
63. Conrad M, Bubb VJ, Schlegel R. The human papillomavirus type 6 and 16 E5 proteins are membrane-associated proteins which associate with the 16-kilodalton pore-forming protein. J Virol. 1993; 67(10):6170-8. [ Links ]
64. Zhang B. The E5 protein of human papillomavirus type 16 perturbs MHC class II antigen maturation in human foreskin keratinocytes treated with interferon-y. Virology. 2003; 310(1):100-108. [ Links ]
65. Ashrafi GH, Haghshenas MR, Marchetti B, O'Brien PM, Campo MS. E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int J Cancer. 2005; 113(2):276-83. [ Links ]
66. Suprynowicz F a, Krawczyk E, Hebert JD, Sudarshan SR, Simic V, Kamonjoh CM, et al. The human papillomavirus type 16 E5 oncoprotein inhibits epidermal growth factor trafficking independently of endosome acidification. J Virol. 2010; 84(20):10619-29. [ Links ]
67. Bouvard V, Matlashewski G, Gu ZM, Storey A, Banks L. The human papillomavirus type 16 E5 gene cooperates with the E7 gene to stimulate proliferation of primary cells and increases viral gene expression. Virology. 1994; 203(1):73-80. [ Links ]
68. Zheng Z-M, Baker CC. Papillomavirus genome structure, expression, and post-transcriptional regulation. Front Biosci. 2006; 11:2286-302. [ Links ]
69. Wang Q, Griffin H, Southern S, Jackson D, Martin A, Mcintosh P, et al. Functional analysis of the human papillomavirus type 16 E1 E4 protein provides a mechanism for in vivo and in vitro keratin filament reorganization. J Virol. 2004; 78(2):821-33. [ Links ]
70. Davy CE, Jackson DJ, Raj K, Peh WL, Southern SA, Das P, et al. Human Papillomavirus Type 16 E1 E4-Induced G 2 Arrest Is Associated with Cytoplasmic Retention of Active Cdk1 / Cyclin B1 Complexes. J Virol. 2005; 79(7):3998-4011. [ Links ]
71. Davy C, McIntosh P, Jackson DJ, Sorathia R, Miell M, Wang Q, et al. A novel interaction between the human papillomavirus type 16 E2 and E1--E4 proteins leads to stabilization of E2. Virology. 2009; 394(2):266-75. [ Links ]

