










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkInvestigaciones Andina
Print version ISSN 0124-8146
Investig. andina vol.14 no.24 Pereira Apr. 2012
FEOCROMOCITOMA BILATERAL: REPORTE DE UN CASO
BILATERAL PHEOCHROMOCYTOMA: CASE REPORT
UM CASO DE FEOCROMOCITOMA BILATERA
Dora Luisa Orjuela Zuluaga*
María Elena Tello Cajiao**
Daniel Andrés Torres Ángel**
* Docente facultad de Ciencias de la Salud, Universidad Tecnológica de Pereira.
** Estudiante de Medicina. Universidad Tecnológica de Pereira.
Resumen
Introducción: el feocromocitoma es una neoplasia endocrina poco común productora de catecolaminas, altamente asociada a riesgo cardiometabólico e hipertensión secundaria. En los últimos años se ha demostrado el incremento de su prevalencia debido al avance diagnóstico en técnicas imagenológicas y bioquímicas, porque la mayoría de estos tumores son descubiertos inicialmente como incidentalomas.
Métodos: se presenta el caso de una paciente, ama de casa de 35 años de edad quien consulta por sintomatología de un año de evolución, consistente en episodios de cefalea, náuseas, palpitaciones, palidez y sudoración.
Resultados: el diagnóstico se apoyó en la detección de altos niveles de catecolaminas en orina y posterior confirmación con estudio imagenológico: ecografía abdominal, TAC contrastado y gammagrafía con MIBG-131.
Conclusión: la paciente fue intervenida quirúrgicamente por adrenalectomía bilateral; se confirmó por anatomía patológica: feocromocitoma bilateral.
Palabras clave: Feocromocitoma; Catecolaminas; Neoplasia Endocrina Múltiple; Hipertensión.
Abstract
Introduction: pheochromocytoma is not a very common endocrine neoplasia which produces catecholamines, highly associated with cardiometabolic and secondary hypertension risks. In recent years, the prevalence of its increase has been demonstrated due to the diagnostic imagenological and biochemical advances, because most of these tumors are initially discovered as incidentalomas.
Methods: the case of a patient is presented, 35 year old homemaker who consults due to symptoms consisting of episodes of cephalea, nausea, palpitations, paleness and sweat over a period of one year.
Results: the diagnose was supported by the detection of high levels of catecholamines in the urine and later confirmation with an imagenological study: abdominal ultrasound scanning, contrasting CAT scan and gammagraphy with MIBG-131.
Conclusion: the patient underwent surgery for bilateral adrenalactomy; it was confirmed by pathological anatomy: bilateral pheochromocytoma.
Keywords: Pheochromocytoma; catecholamines; Neoplasia: Multiple endocrine; Hypertension.
Resumo
Introdução: o feocromocitoma é uma neoplasia endocrina pouco comum, produtora de catecolaminas altamente associada a risco cardio-metabólico e hipertensão secundária. Nos últimos anos se demonstrou o incremento de sua prevalência devido ao avanço diagnóstico em técnicas iconológicas e bioquímicas, porque a maioria desses tumores é descoberta, inicialmente como incidentalomas.
Métodos: apresenta-se o caso de uma dona de casa de 35 anos, que consulta mediante sintomatologia de um ano de evolução, constituída em episódios de cefaleia, náuseas, palpitações, palidez e sudoração.
Resultados: o diagnóstico se apoiou na detecção de altos níveis de catecolaminas na urina e posterior confirmação com estudo iconológico: ecografia abdominal, TAC contrastado e gammagrafia com MIBG-131.
Conclusão: houve intervenção cirúrgica na paciente por adrenalectomia bilateral. Confirmou-se por anatomia patológica que a paciente foi intervida cirurgicamente por: feocromocitoma bilateral.
Palavras chave: Feocromocitoma; Catecolaminas; Neoplasia Endocrina Múltipla; Hipertensão.
Fecha de recibo: Marzo/2011
Fecha aprobación: Octubre/2011
Introducción
El feocromocitoma es un tumor poco común, secretor de catecolaminas, derivado de células cromafines. La primera descripción clínica fue hecha en 1886 en un artículo publicado por el doctor Félix Frankel, donde expuso el caso de una joven de 18 años que murió con los signos clásicos de hipertensión maligna 10 días después de ser ingresada a un hospital en Alemania. (1). En Colombia se encuentran escasos reportes sobre esta condición, pero se sabe que en general puede verse en todos los grupos de edad y sexo, aunque la edad usual de presentación es a los 40 años (2) (3).
En un estudio prospectivo realizado en el Hospital Militar Central y el Hospital de San José en Bogotá, entre los años 1968 y 2007, donde se analizaron 55 casos de patología suprarrenal, se encontró que el feocromocitoma tuvo una frecuencia de aparición de 56,36%, y de esos casos se halló en forma bilateral en solo un paciente (4).
Más del 90% de estos tumores se localizan en las glándulas suprarrenales, y de ellos el 98% son intrabdominales; los extra adrenales se desarrollan en tejido paragangliónico cromafín del sistema nervioso simpático (5). La prevalência de esta neoplasia en la población de hipertensos va de 0,1% al 0,9% y se constituye como una causa importante de hipertensión secundaria (2) (3) (5) (6).
El cuadro clínico característico consiste en episodios de cefalea, palpitaciones, sudoración, hipertensión y taquicardia; sin embargo puede ser asintomático, revelándose como incidentaloma sintomático con crisis hipertensivas y complicaciones cardio o cerebrovasculares (3) (7). Del 25 al 33% está relacionado con mutaciones genéticas específicas, especialmente cuando se presentan bilateralmente, los cuales son frecuentemente hallados como incidentalomas (8). Algunos síndromes relacionados con el feocromocitoma son: la Neoplasia Endocrina Múltiple (MEN I, II), los trastornos neuroectodérmicos como la fibromiomatosis, los Síndromes de Von Hipple- Lindau, de Stuge-Weber y la Triada de Carney (5).
Reporte del caso
Paciente de sexo femenino, 35 años de edad, ama de casa procedente del área urbana de Pereira, quien acudió al Hospital Universitario San Jorge, centro de tercer nivel de atención, para consultar por cefalea global con historia de un año de evolución, 3 a 4 episodios al año de intensidad 8/10, los cuales tenían un tiempo de duración de minutos a horas, síntomas que se exacerban con el esfuerzo, acompañados de náuseas sin vómito, palidez y sudoración. En el último mes refiere mayor intensidad del cuadro con un episodio por semana.
Como antecedentes personales la paciente refirió quemadura en córnea izquierda traumática con trasplante. Padece de artritis reumatoide en tratamiento con hidroxicloroquina y metotrexate. Presentó preeclampsia en su segundo embarazo; tuvo dos gestaciones, una cesárea, un parto, dos vivos. Planifica con método quirúrgico -técnica de Pomeroy- y un ciclo menstrual que dura de 28 a 30 días. Niega hipertensión, diabetes, dislipidemia y tabaquismo. No hay antecedentes familiares de importancia.
En la revisión por sistemas se encontró ausencia de visión por ojo izquierdo desde la intervención quirúrgica por la quemadura. Refirió doloresostoarticulares desde hace 5 años con predominio en manos y en pies. Dice haber tenido palpitaciones ocasionales autolimitadas sin dolor precordial; también dolor abdominal generalizado que cedía espontáneamente, en ocasiones con distensión abdominal; hábito intestinal de cada 3 días con estreñimiento. Cefalea global intensa, acompañada de palidez, sudoración y palpitaciones durante el episodio, síntomas que se exacerbaban con el estrés y el ejercicio; en una ocasión acudió a urgencias encontrándose una presión arterial de 170/115 mmHg, posterior a un afinamiento de presiones, se determinó que no era hipertensa de base. Se concluyó entonces que la paciente padecía de crisis de hipertensión paroxística.
En el examen físico los hallazgos encontrados fueron: pulso de 88 por minuto, presión arterial de 100/60 mmHg, frecuencia cardiaca de 88 frecuencia respiratoria de 18, temperatura de 36.5°C, talla 1.52 y peso 48 kg. La paciente estaba consciente, orientada, hidratada. Se observó opacidad a nivel de ojo izquierdo; en orofaringe tenía prótesis dentaria total superior; en cardiopulmonar se encontraron ruidos cardiacos rítmicos, de buena intensidad, presencia de un soplo sistólico en foco mitral grado II/IV, sin ingurgitación yugular; el murmullo vesicular limpio, no se auscultaron ruidos agregados. El abdomen era blando, depresible, con leve dolor a palpación en flancos, peristaltismo positivo, no se palparon visceromegalias. Refirió artralgias en interfalángicas proximales del 2°, 3° y 4° dedo bilateral de las manos, con presencia de sinovitis en muñeca izquierda. El examen neurológico fue normal, se hizo fondo de ojo sin presencia de papiledema. Las extremidades sin edemas, no hay várices, con pulsos conservados.
Exámenes de laboratorio
Se realizó un abordaje inicial de la paciente con la sospecha de una patología craneoencefálica, por lo cual se solicitaron radiografía y tomografía computarizada de cráneo; se reportaron como normales. (Cuadro 1)
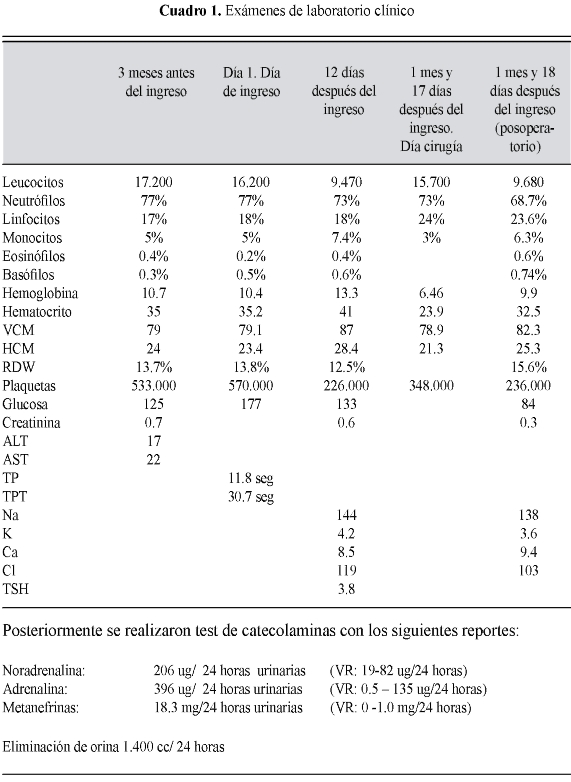
Se realizó ecografía abdominal en donde se aprecian hallazgos en vesícula biliar con múltiples cálculos en su interior; riñones de tamaño y forma normal; a nivel de región suprarrenal derecha se aprecia masa solida de 8x7cm. En región suprarrenal izquierda masa solida de 6.4x7.2 cm, hipervascular. Resto de la ecografía normal.
Se realizó estudio anatómico con TAC abdominal contrastado, en el cual se aprecian dos masas originadas en las glándulas suprarrenales, de manera bilateral, redondeadas, de contornos bien definidos; la derecha es de aspecto sólido con septos en su interior y la izquierda presenta áreas quísticas, sin embargo, es igualmente sólida con septos en su interior, y ambas lesiones captan el medio de contraste de manera importante. Conclusión: masas adrenales bilaterales que sugieren adenomas adrenales, compatible con feocromocitoma bilateral. Resto dentro del límite normal. (Figura 1)
La lectura del electrocardiograma fue normal y reportó hallazgos de prolapso incompetente de la válvula mitral con insuficiencia leve asociada. Fracción de eyección del 71%, resto normal. Gracias a que los resultados paraclínicos confirmaron el diagnóstico de feocromocitoma bilateral, la paciente se sometió a un tratamiento intervencionista tipo quirúrgico: adrenalectomía bilateral.
Preparación antes de la cirugía
Se manejó con medicamentos de segunda línea Catapresán x 150 mg ½ tableta al día, Prazocín x 1 mg media tableta a día, una semana antes de la intervención. En el hospital no se cuenta con el medicamento de elección para manejar la hipertensión causada por feocromocitoma que es Fenoxibenzamina (Bloqueante alfa -adrenérgico) el cual debe iniciarse 4-7 días antes de la cirugía. Adrenalectomía bilateral. Durante el inicio de la cirugía presentó presión arterial 170/110 mmHg y frecuencia cardiaca de 100. Al final de la cirugía presentó presión arterial de 110/70 mmHg y frecuencia cardiaca de 74. La cirugía tuvo una duración aproximada de 3 horas y media.
Manejo post operatorio
Se administraron corticosteroides, líquidos endovenosos y antibióticos. Se manejó en UCI con ventilación mecánica y requirió Norepinefrina y transfusión. Al 7° día de su intervención se anexa Fludocortisona (Astonin) una tableta al día.
Seguimiento de la paciente
Los controles a los tres meses de su intervención de metanefrinas, catecolaminas y glicemias fueron normales. No ha vuelto a tener cefalea, ni sudoración ni palpitaciones. El dolor abdominal ha disminuido.
Anatomía patológica
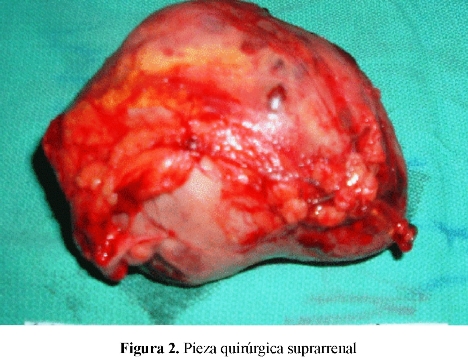
El reporte dado fue suprarrenal derecha: masa de 390 grs. bien delimitada: 10x8x9 cm. pardo marrón de consistencia elástica. Suprarrenal izquierda: masa de 300 grs. bien delimitada, 10.5 x 9.5x 7.5cm pardo marrón consistencia elástica. Conclusión: Suprarrenal derecha: Feocromocitoma. Suprarrenal izquierda: Feocromocitoma. (Figura 2)
Los diagnósticos finalmente considerados fueron feocromocitoma bilateral, diabetes mellitus, colelitiasis, artritis reumatoide, anemia de trastornos crónicos con relación a artritis reumatoide y prolapso de la válvula mitral.
Discusión
El feocromocitoma junto con otros tumores suprarrenales (aldosteronoma y adenoma secretante) se encuentran asociados a riesgo cardiometabólico, por lo cual requieren un manejo adecuado (7). La fisiopatología de la hipertensión arterial secundaria al tumor consta de la alta producción de catecolaminas (generalmente mayores niveles de noradrenalina que adrenalina), que una vez se difunden en la circulación sistêmica actúan principalmente en receptores cardiovasculares y en la activación del sistema nervioso simpático (que a su vez activa la liberación neuronal de catecolaminas), llevando así a estados de hipertensión arterial constante o paroxística, gatillada por situaciones como dolor, emociones, postura y anestesia entre otros, aunque el cuadro clínico individual depende de la cantidad de hormona secretada, del patrón de liberación y de la susceptibilidad individual (9).
Una cuarta parte de los feocromocitomas son detectados premortem y muchos pacientes mueren por complicaciones cardiovasculares antes del diagnóstico. A pesar que la mayoría de los pacientes presentan la triada sintomática característica, es difícil de identificar por su cualidad paroxística que en ocasiones se considera como un simple malestar (7).
La mayoría de feocromocitomas son esporádicos, de localización unilateral y aunque se pueden presentar a cualquier edad son más probables después de los 60 años; cerca de 25-33% de estos tumores se encuentran relacionados con mutaciones genéticas que hacen parte de síndromes hereditarios como el de neoplasia endocrina múltiple 1 y 2 (MEN 1 y 2), el Von Hippel Lindau (VHL), paragangliomas (PGL 4,3 y 1) y neurofibromatosis tipo 1 (NF 1); cada uno consecuencia de mutaciones (autosómicas dominantes) de los genes RET, VHL, SDH (B, C y D) Y NF 1 respectivamente (3)(9).
El tipo de mutación genética específica brinda mayores o menores probabilidades cuando el tumor en cuestión es maligno (10). En el 26-35% de los casos se trata de tumor maligno y en casos esporádicos la prevalencia de malignidad alcanza el 9% (9).
Los feocromocitomas más agresivos son los asociados con las mutaciones del gen SDHB, y cerca del 35% de estos pacientes desarrollan malignidad. Los asociados a síndromes MEN y VHL son los menos metastásicos (8) (9). Es de anotar que ni invasión de cápsula o vasos sanguíneos, ni presencia de mitosis, ni grado de atipias son criterios de malignidad; solo la presencia de metástasis regionales o a distancia (17).
A pesar que el test genético es fundamental, muchos expertos están de acuerdo en realizarlo a pacientes que cumplan los criterios clínicos concernientes a una determinada mutación de la línea germinal; también es importante llevarlo a cabo en pacientes adultos jóvenes, especialmente por la posible relación con VHL y en casos de localización bilateral (9).
En el 10% de los pacientes se puede presentar la forma bilateral, la cual es más común en mujeres y los casos son más frecuentes en los síndromes VHL y MEN; la mutación encontrada principalmente es la Cys634Arg del gen RET. En un paciente con sospecha de bilateralidad es indispensable estudio imagenológico con TAC (tomografía computarizada) y adicionalmente gammagrafía con MIBG (metayodobenzilguanidina) (5)(9)(11).
Cuando se halla incidentaloma bilateral, es importante descartar metástasis, hiperplasia adrenal congénita, adenoma cortical, enfermedades infiltrativas y desarrollar un adecuado diagnóstico diferencial imagenológico, de acuerdo al fenotipo de la imagen (12).
Debido a que los feocromocitomas son un grupo heterogéneo de tumores secretantes con metabolismo variable, es recomendable realizar múltiples pruebas diagnósticas con el fin de aumentar la sensibilidad y especificidad. El abordaje diagnóstico ideal luego de la sospecha clínica es por medio de estudios bioquímicos, y los de elección son la cuantificación de catecolaminas y metanefrinas tanto en plasma como en orina y estudios imagenológicos que ayuden a localizar el tumor descartando focos metastásicos, entre ellos los estudios anatómicos: TAC y RMN (resonancia magnética) y los funcionales: gammagrafía con MIBG y PET (tomografía por emisión de positrones) (2) (8) (9) (13). La RMN tiene una sensibilidad del 93 - 100% superior al TAC cuya sensibilidad es del 85 - 94%; respecto a imagen funcional la PET es superior a la gammagrafía con MIBG; sin embargo, la PET no es muy popular actualmente en nuestro medio (7)(10)(14).
Respecto a tamizaje, el test diagnóstico preferido son las metanefrinas libres en plasma con una sensibilidad del 99% y especificidad del 89%. (6) Se espera en un futuro que el PET con FDOPA (fluor-dihidroxifenilalanina) y/o FDA (fluordopamina) reemplacen el diagnóstico por MIBG. El diagnóstico por gammagrafía MIBG-123 solo ha sido superior al diagnóstico por gammagrafía MIBG-131 en feocromocitomas extraadrenales (10).
Es característico evitar los procedimientos invasivos para su diagnóstico, porque cualquier contacto con el tumor puede precipitar arritmias cardiacas y crisis hipertensivas; por eso la biopsia por aspirado con aguja está contraindicada, además de las altas tasas de hemorragia (5) (12).
La extracción quirúrgica es el paso a seguir después del diagnóstico por localización; el método de elección es adrenalectomía laparoscópica (4)(15). Es fundamental manejar la hipertensión arterial y las posibles complicaciones cardiovasculares, por lo cual es recomendable el uso preoperatorio de un alfa-bloqueante como lo es la fenoxibenzamina 10 mg dos veces al día 14 días antes, con ajustes de la dosis de acuerdo a la respuesta del paciente; además el uso de antagonistas de receptores alfa como lo son el prazocín o doxazocín, betabloqueadores o calcioantagonistas según demanda, y es importante la expansión de volumen antes y después de la cirugía para evitar la hipotensión posoperatoria. Se espera la normalización de los niveles de catecolaminas tanto en plasma como en orina 10 días después de la operación (9) (16).
REFERENCIAS
1. Neumann H, Vortmeyer A, Schmidt D, Werner M, Erlic Z, Cascon A, Bausch B, Januszewicz A, Eng C. Evidence of MEN-2 in the Original description of Classic Pheochromocytoma. N Engl J Med 2007; 357: 1311-5. [ Links ]
2. De La Hoz J. Feocromocitoma. Rev. Col Cir 1994; 9 (3): 225 -238. [ Links ]
3. Fauci A, Braunwald E, Kasper D, Hauser S, Longo D, Jameson JL, Loscalzo J, Eds. Harrison's Principles of Internal Medicine, 17th ed. The McGraw-Hill Companies, Inc. All Rights Reserved 2008. [ Links ]
4. Guzmán Charry JA, Fernández Moncaleano G. Evolución del diagnóstico y el tratamiento de la patología quirúrgica suprarrenal: Serie de 55 casos. Urol. Colomb. Vol. XVIII, No. 1: pp 27-32, 2009. [ Links ]
5. Blake M, Kalra M, Maher M, Sahani D, Sweeney A, Mueller R, Hahn P, Boland G. Pheochromocytoma: An Imaging Chameleon. Radiographics October 2004 24:S87-S99; doi:10.1148/rg.24si045506. [ Links ]
6. Pisoni R, Ahmed MI, Calhoun DA. Characterization and Treatment of Resistant Hypertension. Curr Cardiol Rep. 2009 November; 11(6): 407-413. [ Links ]
7. Ridho FE, Adam FMS, Adam JFM. Adrenal incidentaloma. Acta Med Indones-Indones J Intern Med 41(2):87-93(2009). [ Links ]
8. Havekes B, King K, Lai E W, Romijn J A, Corssmit E PM, Pacak K. New Imaging Approaches to Pheochromocytomas and Paragangliomas. Clin Endocrinol (Oxf). 2010 February; 72(2): 137-145. doi:10.1111/j.1365-2265.2009.03648.x. [ Links ]
9. Karagiannis A, Mikhailidis DP, Athyros VG, Harsoulis F. Pheochromocytoma: an update on genetics and management. Endocr Relat Cancer. 2007 December; 14(4): 935-956. doi: 10.1677/ERC-07-0142. [ Links ]
10. Havekes B, Lai EW, Corssmit EP, Romijn JA, Timmers HJ, Pacak K. Detection and treatment of pheochromocytomas and paragangliomas: current standing of MIBG scintigraphy and future role of PET imaging. Q J Nucl Med Mol Imaging. 2008 Dec; 52(4):419-29. [ Links ]
11. Rodríguez JM, Balsalobre M, Ponce JL, Ríos A, Torregrosa NM, Tebar J, Parrilla P. Pheochromocytoma in MEN 2A Syndrome. Study of 54 Patients. World Journal of Surgery Volume 32, Number 11, 2520-2526, DOI: 10.1007/s00268-008-9734-2. [ Links ]
12. Young WF. The Incidentally Discovered Adrenal Mass. N Engl J Med 2007; 356:601-610 [ Links ]
13. Rocha ME, González G, Tapia JL, Quintero C, Villasmil M, Uzcátegui EC, Ferreira E. Feocromocitoma suprarrenal derecho: reporte de un caso y revisión de la literatura. Rev. venez. Cir; 60(4):173-176, dic. 2007. [ Links ]
14. Sánchez Turcios RA. Feocromocitoma: Diagnóstico y tratamiento. Rev. Mex. Cardiol 2010; 21 (3): 124-137. [ Links ]
15. Walz MK, Alesina PF, Wenger FA, Koch JA, Neumann HP, Petersenn S, Schmid KW, Mann K. Laparoscopic and retroperitoneoscopic treatment of pheochromocytomas and retroperitoneal paragangliomas: results of 161 tumors in 126 patients. World journal of surgery 2006; 30(5):899-908. [ Links ]
16. Sociedad Colombiana de Cardiología y Cirugía Cardiovascular. Guías Colombianas para el diagnóstico y tratamiento de la hipertensión arterial. Revista Colombiana de Cardiología Febrero 2007 Vol. 13 Suplemento 1 ISSN 0120-5633. [ Links ]
17. Álvarez Tostado R, Álvarez Tostado R, Portela Ortiz JM, Olvera Barraza C, Burgos Zuleta A. Feocromocitoma. Presentación de un caso y revisión de la literatura. Asociación Mexicana de Cirugía Endoscópica, A.C.Vol.8 No.3 Jul.-Sep., 2007 pp 148-156 [ Links ]

