










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkInvestigaciones Andina
versión impresa ISSN 0124-8146
Investig. andina vol.15 no.26 Pereira abr. 2013
Diagnóstico de la deficiencia de hidroxi-acil CoA deshidrogenasa de cadena larga, mediante la incubación de fibroblastos con ácido [9,10)(n)-3h] palmítico y ácido [9,10)(n)-3h] mirístico
Diagnosis Long-chain hydroxy acyl-CoA dehydrogenase deficiency, by fibroblasts incubation with [9, 10)(N)-3H] palmitic and [9,10)(N)-3H] miristic acids
Diagnóstico da deficiência de hidroxi-acil CoA deshidrogenasa de longa cadeia, mediante incubação de fibroblastos com ácido [9,10)(n)-3h] palmítico e ácido [9,10)(n)-3h] mirístico
José Henry Osorio
* Universidad de Caldas, Manizales, Colombia. jose.osorio_o@ucaldas.edu.co
Resumen
Introducción: la deficiencia de hidroxi-acil CoA deshidrogenasa de cadena larga, es una enfermedad metabólica autosómica recesiva, caracterizada por acidosis, hipoglicemia, cardiomiopatía, daño hepático y rabdomiólisis. Objetivo: analizar las tasas de oxidación de sustratos tritiados por fibroblastos de pacientes con deficiencia de hidroxi-acil-CoA deshidrogensa y controles. Métodos: fibroblastos de pacientes y controles se incubaron con [3H]-palmitato y [3H]-miristato y se determinó la oxidación de los mismos en nmol/h/mg proteína. Resultados: se encontró deficiente la oxidación de sustratos tritiados por parte de los fibroblastos procedentes de los pacientes que presentaban la deficiencia, con una tasa de oxidación de 43 y 48% para palmitato y miristato respectivamente. Conclusión: esta técnica modificada permite el diagnóstico in vitro de la deficiencia de hidroxi-acil-CoA de cadena larga.
Palabras Clave: ß-Oxidacion; Ácidos Grasos; Metabolismo.
Abstract
Introduction: long-chain hydroxy acyl-CoA dehydrogenase (LCHAD) deficiency, is an autosomal recessive metabolic disease, characterised by acidosis, hypoglycaemia, cardiomyopathy, liver damage, and rhabdomyolysis. Objective: to analyse the oxidation rate of tritiated substrates in fibroblasts of patients suffering the deficiency and controls. Methods: fibroblasts from patients and controls were incubated with [3H]-palmitate and [3H]-miristate and the oxidation of these substrates were measured in nmol/ hour/mg protein. Results: it was found depressed oxidation of tritiated substrates in fibroblasts from patients suffering the deficiency, with a oxidation rate of 43% and 48% for palmitate and miristate respectively. Conclusion: this modified technique enables us the in vitro diagnosis or long-chain hydroxy acyl-CoA dehydrogenase.
Keywords: ß-Oxidation; Fatty Acids; Metabolism.
Resumo
Introdução: a deficiência de hidroxi-acil CoA deshidrogenasa de longa cadeia, é uma doença metabólica autossômica recessiva, caracterizada por acidose, hipoglicemia, cardiomiopatia, dano hepático e rabdomiólise. Objetivo: analisar as taxas de oxidação de substratos tritiados por fibroblastos de pacientes com deficiência de hidroxi-acil-CoA desidrogenas e controles. Métodos: fibroblastos de pacientes e controles se incubaram com [3H]-palmitato e [3H]-miristato e se determinou a oxidação dos mesmos em nmol/h/mg proteína. Resultados: encontrou-se deficiente a oxidação de substratos tritiados por parte dos fibroblastos procedentes dos pacientes que apresentavam a deficiência, com taxa de oxidação de 43 e 48% para palmitato e miristato respectivamente. Conclusão: esta técnica modificada permite o diagnóstico in vitro da deficiência de hidroxi-acil-CoA de longa cadeia.
Palavras Chave: ß-Oxidacäo; Ácidos Gordurosos; Metabolismo.
Fecha de recibo: Julio/2012
Fecha aprobación: Diciembre/2012
Introducción
La ß-oxidacion mitocondrial es el proceso mayoritario de oxidación de los ácidos grasos, fuente energética de gran importancia tanto para el músculo esquelético como para el músculo cardiaco, principalmente durante los períodos de ejercicio prolongado y el ayuno.
Una vez al interior de la mitocondria, los esteres acil-CoA de los ácidos grasos son degradados a través de cuatro reacciones secuenciales a saber:
a. Reducción en la posición a-ß del acil-CoA produciendo 2,3,-enoil-CoA;
b. Hidratación del doble enlace dando lugar a especies estereosespecíficas, L-3- hidroxiacil-CoA.
c. Oxidación en la posición 3-hidroxi, lo cual da lugar a 3-cetoacil-CoA.
d. Rotura tiolítica del 3-cetoacil-CoA a acetil-CoA y acil-CoA, cuya cadena carbonada es ahora dos átomos de carbono más corta.
Este acil-CoA puede volver a entrar en el ciclo de la ß-oxidación tantas veces como sea necesario, hasta su completa oxidación a acetil-CoA. Se ha descrito un complejo de la membrana interna mitocondrial (llamado 'proteína trifuncional' TFP) que realiza las tres actividades enzimáticas: enoil-CoA hidratasa, hidroxiacil-CoA deshidrogenasa y cetoacil-CoA tiolasa, pasos b, c, y d de la ß-oxidacwn mitocondrial, para los ácidos grasos de cadena larga (1).
La deficiencia de 3-hidroxiacil-CoA deshidrogenasadecadenalarga(LCHAD), sola, o como parte de la deficiencia de proteína trifuncional (TFP), se manifiesta típicamente durante los episodios de ayuno o enfermedad, como hipoglicemia hipocetócica. Las complicaciones agudas o crónicas de la deficiencia de LCHAD pueden ser manejadas evitando el ayuno y consumiendo una dieta baja en ácidos grasos de cadena larga y muy larga, así como suplementando a los pacientes con ácidos grasos de cadena media y corta (2). La deficiencia de LCHAD es escasa y está asociada con alteraciones durante el embarazo (3-5), retinopatías (6) y otras manifestaciones clínicas (7), y puede ser causada por mutaciones en la subunidad alfa (OMIM # 600890) o en la subunidad beta (OMIM #143450) de la TFP, siendo la mutación de cambio de sentido c.1528G>C en la subunidad alfa, la que más prevalece (8,9).
El presente estudio analizó la oxidación de sustratos tritiados en fibroblastos de pacientes con deficiencia de LCHAD, como herramienta diagnóstica para este tipo de alteraciones metabólicas.
Materiales y métodos
Estudio de tipo experimental descriptivo. El material biológico empleado ha sido fibroblastos de 2 pacientes, cedidos por la Sección de Errores Congénitos del Metabolismo (IBC), Servicio de Bioquímica y Genética Molecular, Hospital Clínic de Barcelona, con deficiencia de LCHAD. Esta deficiencia fue confirmada por estudios enzimáticos, moleculares o ambos. Como controles se utilizaron 20 cultivos diferentes de fibroblastos normales.
Células utilizadas: fibroblastos de pacientes y controles (4-20 pasajes), fueron cultivados en bicarbonate-HEPES-buffered Eagle's minimal essential médium (MEM), suplementado con 10% (v/v) newborn calf serum, y 1% (v/v) de antibiótico (gentamicina) a 370C, en estufa con 5%CO2/95% de aire. Después de alcanzar el punto de confluencia (80-100%), las células fueron lavadas dos veces con PBS y la solución se tripsinizó (1 ml de tripsina-EDTA a 370C) y posteriormente se neutralizó mediante la adición de 3-5 ml de MEM (Minimun EsentialMedium). Las células fueron transferidas y centrifugadas a 337 X g (5 min, 20oC) en tubos cónicos de 10 ml (0.8-1.2 mg proteína), quedando listas para ser incubadas en presencia de sustratos tritiados. Se utilizó el método de Lowrry et al. (10) para la determinación de la proteína.
Preparación de sustratos tritiados: el método utilizado fue el de Manning (11) con modificaciones, y la secuencia metodológica para obtener las mezclas radioactivas fue la siguiente:
Solución A: Ácido palmítico 12.5 mg/1 ml de etanol al 95% o ácido mirístico 12.5 mg/1 ml etanol 95%;
Solución B: Albúmina 25 mg/ml = 75 mg/30 ml de PBS 1 Reactivo C: ácido [9,10)(n)-3H] palmítico (Diluido en tolueno, 50-62 mCi/mmol, 1 mCi/ ml) (Amersham) o ácido [9,10)(n)-3H] mirístico (Diluido en tolueno, 40-60 Ci/ mmol, 1mCi/ml) (Amersham). Se mezcla en un tubo plástico de 3 ml, 25 µL de A+3.8 µL de C (3.8 µCi), y el solvente se evapora bajo gas nitrógeno. Se agregan 2.5 ml de B y se analiza la mezcla en el contadorde centelleo (LS3801-Beckman), (50 µl de la solución+10 ml de líquido de centelleo (Ready Safe). Luego la mezcla es llevada al baño de ultrasonido durante 15 minutos y posteriormente a incubación al baño a 370C durante 40 min, para luego llevarla nuevamente al baño de ultrasonido durante 30 min, y se centrifuga durante 20 min a 5000 rpm. Se traspasa el máximo de volumen sobrante a otro tubo plástico de 3 ml, y se analizan 50 µL de la solución, como se describió anteriormente.
Preparación de las columnas de intercambio iónico: pueden ser usadas las resinas Dowex 1X8-200 o Dowex 1X2-400, se le agrega agua destilada a la resina hasta que se hidrate. Se sellan al mechero pipetas Pasteur, aproximadamente a 3 cm de la punta, y se aísla el cuerpo de la pipeta de la punta, mediante la introducción de algodón comprimido, tratando que el volumen de algodón ocupe aproximadamente 1 cm.
Se toma igual volumen de agua que de resina, se lleva a agitación suave y mientras se agita, se toman 2.5 ml y se depositan cuidadosamente en la pipeta, humedeciendo previamente el algodón con agua milli Q, para evitar la retención de burbujas de aire que puedan posteriormente hacer caminos en la resina. Es recomendable conservar las pipetas en posición vertical.
Después de depositar la resina se debe verificar que no queden burbujas de aire y que la pipeta no pierda agua, para que la resina permanezca hidratada; luego las columnas pueden ser conservadas en refrigeración hasta su utilización.
Evaluación de la oxidación de sustratos tritiados por los fibroblastos: para evaluar la oxidación de [3H]-palmitato por los fibroblastos, se toma el pellet de células previamente resuspendido en 500 µL de PBS 1. Se utilizan bandejas de incubación con capacidad para 24 pozos. El blanco contiene 40 µL (0.05µCi) de mezcla radioactiva y 160 µL de PBS 2; las muestras contienen 60 µL de células resuspendidas, 40 µL (0.05 µCi) de mezcla radioactiva, y 100 µL de PBS 2. La bandeja se envuelve en papel de aluminio y se incuba a 370C durante 4 horas en cámara de CO2 5%, aire 95%. La reacción se detiene, colocando la bandeja en hielo y agregando a cada pozo 200 µL de TCA 10%. El contenido de los pozos se traspasa a los tubos para microcentrífuga y se centrifugan a 5000 rpm durante 5 min. 360 µL de la mezcla son transferidos de nuevo a un tubo de microcentrífuga diferente, que contiene 50 µL de NaOH 1 M. Esta mezcla se mantiene en hielo hasta el momento de aplicar a la columna. Para la aplicación a la columna de intercambio iónico, la punta sellada de la pipeta Pasteur (columna), se parte cuidadosamente, se coloca un vial bajo la pipeta y se deja escurrir su contenido, aplicando luego cuidadosamente 455 µL de mezcla de reacción a la columna, dejando escurrir nuevamente. Para el goteo se lava la columna tres veces con 500 µL agua milli Q, recogiendo todo el producto del lavado. Se desecha la columna y se agrega a cada vial 10 ml de líquido de centelleo, agitando fuertemente para su posterior análisis.
De acuerdo con el artículo 11 literal a. de la Resolución No. 8430 de 1993 del Ministerio de Salud de Normas Científicas, Técnicas, y Administrativas para la Investigación en Salud, el presente estudio es considerado sin riesgo.
Resultados
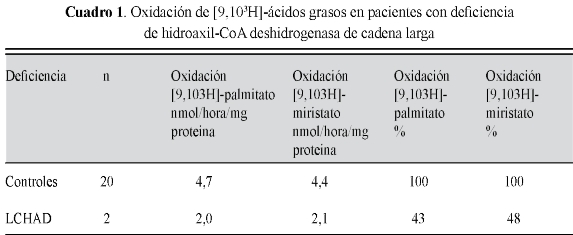
La oxidación de palmitato y miristato tritiados en nmol/hora/mg proteína y el porcentaje de oxidación de los sustratos tritiados por parte de los fibroblatos de los pacientes comparados con los controles paralelos, se halló muy deprimida. Los porcentajes de oxidación son similares tanto para el palmitato como para el miristato tritiados (43-48% comparado con los controles: 100%). (Cuadro 1)
Discusión
El diagnóstico de la deficiencia de hidroxiacil-CoAdeshidrogenasadecadena larga, se basa en las manifestaciones clínicas, principalmente en hipoglicemia, hepatomegalia, cardiomiopatía, trastornos del ritmo cardíaco, debilidad muscular o miopatía. También se han reportado síndrome de muerte súbita del lactante, retinitis pigmentosa, neuropatía periférica y mioglobinuria. Las investigaciones bioquímicas demuestran niveles bajos de carnitina en sangre, así como niveles elevados de C14:1n-9 en suero y prominencia de hidroxiacilcarnitinas en sangre (C16OH, C18OH,C18:1OH) (12).
Los estudios 'in vitro' de estos pacientes incluyen entre otros la utilización de fibroblastos y linfocitos (13). En los fibroblastos se encuentran preservadas las relaciones entre la oxidación de ácidos grasos, los sistemas de transferencia de electrones y otras vías que interactúan en el metabolismo intermediario. La integración de estos sistemas es esencial para el estudio de la organización y control de las enzimas involucradas en la ß-oxidación mitocondrial, lo que hace este tipo de células ideal para el presente estudio. A partir del desarrollo de los ensayos para medir las tasas de oxidación de ácidos grasos en células mediante la incubación con substratos radioactivos, se avanzó notablemente en el diagnóstico de las deficiencias de la ß-oxidación mitocondrial de los ácidos grasos. Moon y Rhead desarrollaron en 1987 (14), un método de valoración de 3H2O en fibroblastos con ácidos grasos tritiados. La liberación de tritio unido a los carbonos 9 y 10 del palmitato depende de tres mecanismos:
(a) la acción de una acyl-CoA deshidrogenasa formando un 2,3-enoyl-CoA ester y transfiriendo el 50% del tritio a la proteína de transmisión de electrones (FADH);
(b) la reacción de la 3-hidroxiacil-CoA deshidrogenasa que remueve la mitad del tritio remanente, el cual finaliza en el NADH; y
(c) el ciclo del ácido tricarboxílico, el cual libera el 25% restante. La incorporación final del tritio al agua es por lo tanto dependiente de la reoxidación de esos cofactores en la cadena de transporte de electrones.
Los complejos II, III y IV de la cadena respiratoria, son necesarios para captar tritio que viene de la proteína de transferencia de electrones, y los complejos I, III y IV son precisos para la re-oxidación del NADH, por lo cual la prueba puede ser utilizada también en ciertos defectos de la cadena respiratoria (15). Manning y colaboradores (11) postularon que [9,10)(n)-3H]-el miristato posee claras ventajas sobre [9,10-3H]-el palmitato para la detección de trastornos tales como la deficiencia de acil-CoA deshidrogenasa de cadena media MCAD, donde la B-oxidación se encuentra detenida a nivel de los sustratos con 8 átomos de carbono. Cabe mencionar que estos autores trabajaron con células de pacientes que sufrían deficiencias de MCAD y MAD, mientras que este trabajo se desarrolló con células de pacientes con deficiencia de LCHAD.
Otros estudios que utilizan esta técnica, reportan niveles de oxidación de ácidos grasos en varias deficiencias de esta vía (16, 17). En este estudio se encontró para esta deficiencia una tasa de oxidación que oscila entre el 43 y el 48% comparada con los normales; al igual que otros autores lo hicieron con otras deficiencias (18).
Es posible concluir que la valoración de agua tritiada es un buen método para confirmar una deficiencia de hidroxi acil-CoA deshidrogenasa de cadena larga, porque es una técnica de fácil acceso en varios laboratorios, no así procesos como la espectrometría de masas en tándem. En el presente estudio fue evaluada la oxidación de sustratos lipídicos en células de pacientes con deficiencia de LCHAD, mientras que autores como Manning y col. (11), trabajaron solamente con deficiencias de MCAD y MAD.
Los resultados de esta investigación expresan la aplicabilidad de esta práctica para la deficiencia de hidroxi-acil CoA deshidrogenasa de cadena larga, mediante la incubación de fibroblastos con ácido [9,10)(n)-3h] palmítico y acido [9,10)(n)-3h] mirístico.
REFERENCIAS
1. Chegary, M. Brinke, H. Ruiter, J.P. Wijburg, F.A. Stoll, M.S. Minkler, P.E. et al. (2009). Mitochondrial long chain fatty acid beta-oxidation in man and mouse. Biochim Biophys Acta; 1791(8):806-15. [ Links ]
2. Gillingham, M. Van Calcar, S.C. Ney, D.M. Wolv, J. Harding, C.O. (1999). Dietary Management of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD). A case report and survey. J Inher Metab Dis; 22:123-31. [ Links ]
3. Ibdah, JA. (2006). Acute fatty liver of pregnancy: an update on pathogenesis and clinical implications. World J Gastroenterol;12(46):7397-404. [ Links ]
4. Jebbink, J. Wolters, A. Fernando, F. Afink, G. van der Post, J. Ris-Stalpers, C. (2012). Molecular genetics of preeclampsia and HELLP syndrome. Biochim Biophys Acta;1822(12):1960-9. [ Links ]
5. Griffin, A.C. Strauss, A.W. Bennett, M.J. Ernst, L.M. (2012). Mutations in long-chain 3-hydroxyacyl coenzyme a dehydrogenase are associated with placental maternal floor infarction/massiveperivillous fibrin deposition. Pediatr Dev Pathol; 15(5):368-74. [ Links ]
6. Fletcher, A.L. Pennesi, M.E. Harding C.O. Weleber, R.G. Gillingham, MB. (2012) Observations regarding retinopathy in mitochondrial trifunctionalprotein deficiencies. Mol Genet Metab;106(1):18-24. [ Links ]
7. Spiekerkoetter, U. (2012). Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J Inherit Metab Dis; 33(5):527-32. [ Links ]
8. Saudubray, J.M. Martin, D. De Lonlay, D. Touati, P. Poggi-Travert, G. Bonnet, F. et al. (1999). Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis; 22:488-502. [ Links ]
9. Eskelin, P.M.A. Laitinen, K.A. Tyni, T.A. (2010). Elevated hydroxyacylcarnitines in a carrier of LCHAD deficiency during acute liver disease of pregnancy - A common feature of the pregnancy complication?. Mol Genet Metab;100(2):204-06. [ Links ]
10. Lowry, O.H. Rosebrough, N.J. Farr, A.L. Randall, R.J. (1951). Protein measurement with the Folinphenol reagent. J Biol Chem;193:265-75. [ Links ]
11. Manning, N.J. Olpin, S.E. Pollit, R.J. Webley, J.A. (1990). Comparison of 9.10-3HPalmitic and 9.10-3Hmyristic acids for the detection of defects of fatty acid oxidation in intact cultured fibroblasts. J Inher Metab Dis.; vol 13:58-68. [ Links ]
12. Martínez-Quintana, E. Peña-Quintana, L. Artíles-Vizcaíno, J.A. Rodríguez-González, F. (2009). Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency and cardiogenic shock. International J. Cardiol; 136: e1-e2. [ Links ]
13. Gillingham, M.B. Scott, B. Elliott, D. Harding, C.O. (2006). Metabolic control during exercise with and without medium-chain triglycerides (MCT) in children with long-chain 3-hydroxy acyl-CoAdehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol Genet Metab; 89:58-63. [ Links ]
14. Moon, A. Rhead, W.J. (1987). Complementation analysis of fatty acid oxidation disorders. J Clin Invest;79:56-94. [ Links ]
15. Venizelos, N. Von Dobeln, U. Hagenfeld, L. (1998). Fatty acid oxidation in fibroblasts from patients with defects in ß-oxidation and in the respiratory chain. J Inher Metab Dis; 21:409-15. [ Links ]
16. Nada, M.A. Rhead, W.J. Sprecher, H. Schulz, H. Roe, C.R. (1995). Evidence for intermediate channeling in mitochondrial ß-oxidation. J Biol Chem; 270:530-35. [ Links ]
17. Ventura, F.V. Costa, C.G. Struys, E.A. Ruiter, J. Allers, P. Ijlst, L. et al (1999). Quantitative acylcarnitine profile in fibroblasts using U-13Cpalmitic acid: an improved tool for the diagnosis of fatty acid oxidation defects. Clin Chem Acta; 281:1-17. [ Links ]

