










Serviços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Facultad Nacional de Agronomía Medellín
versão impressa ISSN 0304-2847
Rev. Fac. Nac. Agron. Medellín vol.69 no.2 Medellín jul./dez. 2016
https://doi.org/10.15446/rfna.v69n2.59133
DOI: http://dx.doi.org/10.15446/rfna.v69n2.59133
Next generation sequence analysis of the forage peanut (Arachis pintoi) virome
Secuenciación de nueva generación del viroma del maní forrajero (Arachis pintoi)
Pablo Andrés Gutiérrez Sánchez1, Helena Jaramillo Mesa1 and Mauricio Marín Montoya1
1 Facultad de Ciencias. Universidad Nacional de Colombia. A.A. 3840, Medellín, Colombia.
Received: January 29, 2016; Accepted: March 18, 2016
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.

ABSTRACT
Forage peanut (Arachis pintoi) is one of the forage crops best adapted to tropical agroecosystems where it is used as ground cover in urban areas and slopes, in the preservation of soils cultivated with coffee, African oil palm and citrus and as animal feed in combination with gramineous plants. A. pintoi is considered to be highly resistant to plagues and diseases; however, in recent years there has been a marked increase of plants showing symptoms typical of viral infection. In this work, Next Generation Sequencing (NGS) was used to confirm the presence of virus in symptomatic A. pintoi plants collected in urban areas in Medellín (Colombia). Transcriptome analysis revealed the presence of 3,291,089 reads related to viruses in the families Potyviridae, Luteoviridae and Alphaflexiviridae and resulted in the complete genome assembly of Peanut mottle virus (9707 nt), Turnip yellows virus (5578 nt) and two variants of a virus with phylogenetic affinity to the genus Allexivirus. These two variants lack ORF6 present in Allexivirus and probably belong to an uncharacterized genus within the family Alphaflexiviridae. The presence of at least three viruses infecting A. pintoi in Colombia highlights the importance of starting a germplasm clean-up program of the plant material used as seed in this crop.
Key words: Alphaflexiviridae, Polerovirus, Potyvirus, Viral genomes
RESUMEN
El maní forrajero (Arachis pintoi) es uno los cultivos forrajeros mejor adaptado a los agroecosistemas tropicales, donde se utiliza en mezclas con gramíneas para alimentación animal, como alternativa para la cobertura del suelo en áreas urbanas y talúdes y para la conservación de suelos en plantaciones de palma africana, café y cítricos. Aunque el maní forrajero se considera una planta tolerante a plagas y enfermedades, en los últimos años se ha observado en Colombia el aumento de síntomas asociados a enfermedades virales. Con el objeto de evaluar la ocurrencia de virus en dichos materiales sintomáticos, en el presente estudio se utilizó la metodología de Secuenciación de Nueva Generación (NGS) del transcriptoma de un grupo de muestras de A. pintoi procedentes de zonas urbanas en Medellín (Colombia). Los resultados indicaron la presencia de 3.291.089 reads asociados a genomas virales de miembros de las familias Potyviridae, Luteoviridae y Alphaflexiviridae; siendo posible obtener los genomas completos del Peanut mottle virus (9707 nt), Turnip yellows virus (5578 nt) y de dos variantes de un virus filogenéticamente relacionado con el género Allexivirus. Estas dos variantes carecen del ORF6 presente en Allexivirus, por lo que posiblemente se trata de miembros de un género hasta ahora no caracterizado en la familia Alphaflexiviridae. La ocurrencia de al menos tres virus que infectan plantas de A. pintoi en Colombia, llama la atención sobre la necesidad de emprender un trabajo de limpieza de germoplasma en el material de siembra utilizado para el establecimiento de este forraje tropical.
Palabras claves: Alphaflexiviridae, Polerovirus, Potyvirus, Genomas virales
Forage peanut (Arachis pintoi Krapov. & Gregory), locally known as 'Mani Forrajero Perenne' is a Leguminosae (section Caulorrhizae) plant native of the states of Goiás, Bahia and Minas Gerais in Central Brazil (Krapovickas and Gregory, 1994; Palmieri et al., 2009). A. pintoi is a diploid perennial herb (2n=20) of tetrafoliolate leaves that can reach heights between 20 to 40 cm and is well adapted for clonal propagation by means of stolons (Rincón et al., 1992; Lavia et al., 2011). This plant is widely used as forage to improve the nutritional quality of pastures and also as ground cover in substitution of common grasses in tropical and subtropical regions of Australia, Bolivia, Brazil, Colombia, Costa Rica, Honduras and Venezuela (Valls, 1996; Palmieri et al., 2010). A. pintoi was introduced in Colombia as part of an investigation aimed at finding tropical legumes adapted to Oxisol savanna conditions. Currently, the most important cultivars is CIAT 17434, which was selected from a set of more than 40 accessions of wild species of the genus Arachis from collections in the United States (University of Florida and USDA) and Australia (CSIRO) (Rincón et al., 1992).
Forage peanut has a high nutritive value (13-25% crude protein, 60-70% dry matter digestibility), low levels of condensed tannins, and is well adapted to acid and low fertility soils (Lascano and Thomas, 1988). Moreover, A. pintoi has good legume dry matter yields, can withstand heavy grazing conditions and is compatible with mat-forming species such as Brachiaria humidicola and B. dictyoneura (Rincón et al., 2001). A. pintoi is resistant to a wide range of pests and diseases; however, there are reports of fungal infections with Cercospora sp., Phomopsis sp., Periconia sp., Cylindrocladium sp., Colletotrichum gloeosporioides, Sphaceloma arachidis and Rhizoctonia solani, none of which causes serious damage (Rincón et al., 1992). Even though some studies have reported the presence of yellowing, ringspots and mottle symptoms, there are few investigations regarding viruses infecting A. pintoi. The only work available dates from 1991 and described the natural infection of A. pintoi affected by a potyvirus related to Peanut mottle virus (PeMoV) in various localities in the province of Valle del Cauca in Colombia (Morales et al., 1991). Electron microscopy analysis of leaves exhibiting ringspot symptoms demonstrated the presence of filamentous flexuous particles of about 750 nm in length and 15 nm in diameter that induced the formation of cytoplasmic cylindrical inclusions (Morales et al., 1991). PeMoV, is transmitted non-persistently by aphids and seeds in peanut (Araquis hypogaea) and can also infect soybean and pea. In 2006, this virus was also found in rhizoma peanuts (Arachis glabrata), a forage crop with increasing acreage (>10,500 ha) in the coastal plain region of the United States, causing chlorotic ringspots (Nischwitz et al., 2007).
With the commercial availability of Next-Generation sequencing platforms, the strategies for detecting viruses changed dramatically as it is now possible to obtain detailed genome information without previous knowledge of specific sequences and at very low cost (Rossinck et al., 2015). Currently, the systems most widely used are 454 GS FLX+ (Roche), HiSeq2000, 2500 and MiSeq (Illumina) and SOLiD (ABI), which differ in their sequencing principles and methods of sample preparation. The illumina HiSeq and MiSeq platforms are characterized by the generation of very high throughput of short mate-paired-end reads generated by a bridge PCR that can result in millions of sequence clusters in a flow-cell. In this platform, reads of approximately 100 bp are obtained by synthesis using fluorescently-labeled nucleotides, resulting in about 1 Tb of sequence data per run (Adams et al., 2009; Wu et al., 2015). Using NGS in the last five years at least 50 plant viruses have been discovered, 36 of which were classified into new families and nine were chosen as type species for an equal number of genera (Wu et al., 2015). In Colombia, the NGS study of plant viromes allowed the identification and characterization of new virus species, most of them infecting solanaceous crops of economic importance such as Tamarillo leaf malformation virus (TaLMV), Potato virus Y (PVY), Potato virus V (PVV), Potato yellow vein virus (PYVV), Potato virus X (PVX), Potato virus S (PVS) and Andean potato latent virus (APLV) (Kreuze et al., 2013; Villamil-Garzón et al., 2014; Gutiérrez et al., 2014a, 2014b, 2016).
Close to 31 viruses representing 14 genera have been reported to naturally infect groundnut in different countries, including Tomato spotted wilt virus (TSWV), Groundnut bud necrosis virus (GBNV), Tobacco streak virus (TSV), Groundnut rosette assister virus (GRAV), Groundnut rosette virus (GRV), satellite RNA associated with GRV and/or GRAV, Peanut clump virus (PCV), Bean common mosaic virus (BCMV), Peanut mottle virus (PeMoV) and Cucumber mosaic virus (CMV) (Sreenivasulu et al., 2008). As a detailed description of viruses infecting A. pintoi is lacking at present, we present a genome sequence analysis of the forage peanut virome in Colombia using Next-Generation Sequencing (NGS).
MATERIALS AND METHODS
This work was performed on a bulk sample of the A. pintoi leaves showing symptoms of leaf yellowing and green vein banding, as well as irregular dark islands and mottling on young leaves. Samples were collected in Medellín (Antioquia, Colombia) (Figure 1). Fresh leaf tissue was ground using liquid nitrogen and RNA extracted with the GeneJET Plant RNA Purification kit (Thermo, EEUU). The integrity of total RNA was determined using a 2100 Bioanalyzer (Agilent Technologies, EEUU). rRNA was depleted with the TruSeq Stranded Total RNA with Ribo-Zero (Illumina, EEUU) while the TruSeq RNA Sample Preparation kit was used for cDNA library construction (Illumina). Sequencing was performed with the Illumina HiSeq 2000 system provided by Macrogen (South Korea). Low-quality bases were trimmed from both ends using the Phred algorithm implemented in the program SeqTk v.r82 (https://github.com/lh3/seqtk).

Preliminary identification of viral sequences was performed with a BLASTN search against a local database containing all plant virus species currently accepted by the ICTV. Potyvirus and Polerovirus where assembled by mapping reads against the complete genomes of PeMoV (gb: NC_002600) and Turnip yellows virus (TuYV) (gb: NC_003743), respectively. The alphaflexivirus genomes were assembled using an iterative Perl routine that used BLASTN to identify reads with 30 nt overlaping segments at both ends of a seed sequence. Genomes were confirmed by de novo reconstruction using Trinity (Grabherr et al., 2011) and mapping with Bowtie2 (Langmead et al., 2009). Open reading frames (ORFs) were identified using BLASTX and ORF finder (Gish and States, 1993). Sequences have been deposited in GenBank under accession codes KU708532 (PeMoV), KU726090-1 (TuYV), KX058345 (Arachis pintoi virus A) and KX058346 (Arachis pintoi virus B).
Phylogenetic reconstruction of amino acid sequences were inferred by the Maximum Likelihood (ML) method using the LG+G+I substitution model (Le and Gascuel, 2008). ML analyses using nucleotide sequences were calculated with the HKY+G+I model (Hasegawa et al., 1985). Sequence alignments were performed with MUSCLE (Edgar, 2004) and models selected with Modeltest (Posada and Crandall, 1998). Evolutionary analyses were conducted in MEGA6 using 1000 bootstrap replicates (Tamura et al., 2013).
RESULTS AND DISCUSSION
NGS analysis of the A. pintoi transcriptome resulted in a paired-end library of 40,712,018 reads for a total of 8,142,403,600 nt. BLASTN analysis indicated that 7.2 percent of the reads (3,291,089) were significantly similar to plant viruses within the Potyviridae, Luteoviridae and Alphaflexiviridae families (Figure 2).
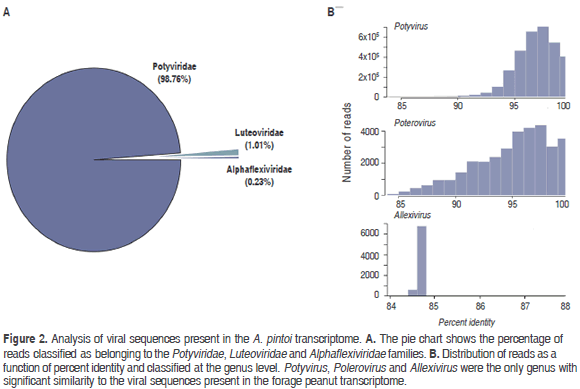
Sequences related to Potyviridae members comprised 98.76 % percent of all viral sequences, which were further classified as belonging to the genus Potyvirus. Reads shared an average 96.67% (84 - 98.02%) percent identity with species of this genus; the most abundant hit corresponded to PeMoV (3,250,148). Reads in the family Luteoviridae shared identities ranging from 84 to 100% (average 94.82%) with members of the genus Polerovirus, the most abundant hit corresponding to TuYV (21,575). A group of 7,593 reads were classified as related to members of the family Alphaflexiviridae with the highest similarity to Shallot virus X (ShVX) (7,521), a species in the genus Allexivirus. However, the low average percent identity observed (84.7%), suggests that these sequences are probably significantly different to currently known members of this family.
Potyvirus
Mapping of the A. pintoi transcriptome to a PeMoV reference sequence resulted in a consensus assembly of 9707 nt (excluding the polyA tail) with an average coverage of 367x encoding a putative polyprotein of 3099 amino acids between positions 122 to 9421 (Figure 3). A BLASTN search using the complete assembly as query against the NCBI database confirmed that the assembled contig was closely related to Peanut mottle virus strain M (gb:AF023848) and Peanut mottle virus isolate Habin (gb:KF977830) with a global percent nucleotide identity of 96% against both sequences. The consensus assembly was named Peanut mottle virus isolate pintoi (PeMoV-pintoi) to highlight the isolation host. In the PeMoV-pintoi assembly, 242 polymorphic positions were identified, with a transition/transversion ratio of 7.96. Within the coding region, most nucleotide changes (159), occurred in the third codon position, in contrast to 41 in the first position and 29 in the second. The following are the 61 amino acid substitutions observed: P1 (10); HC-Pro (10); P3 (10); CI (10); 6K2 (2); NIa-VPg (3); NIa-Pro (3); NIb (10) and CP (3).
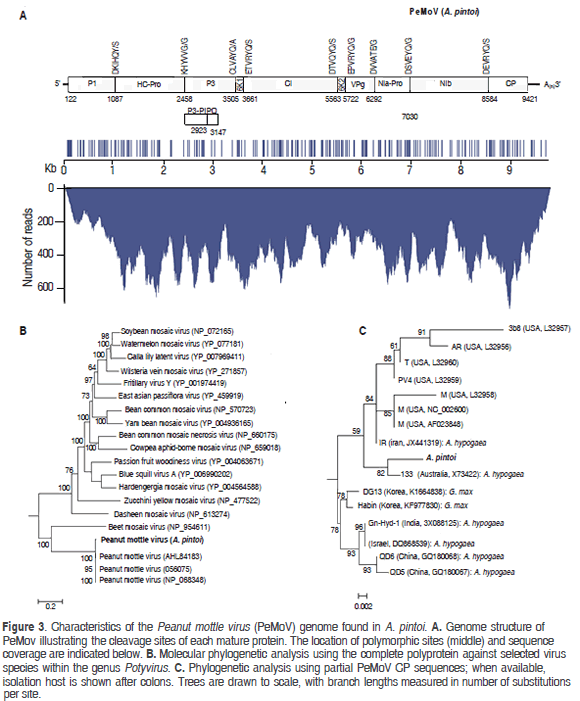
Protease cleavage sites within the polyprotein were identified at amino acid positions 322, 779, 1128, 1180, 1814, 1867, 2057, 2303 and 2821 giving rise to putative mature proteins P1 (36.99 kDa), HC-Pro (51.21 kDa), P3 (40.27 kDa), 6K1(5.72 kDa), CI (70.89 kDa), 6K2 (6.15 kDa), NIa-VPg (21.35 kDa), NIa-Pro (27.72 kDa), Nib (59.75 kDa) and CP (31.53 kDa). P1 and HC-Pro are predicted to be processed autocatalitically at recognition sequences DKIHQY/S and KHYVVG/G in agreement with previous reports (Adams et al., 2005). The fusion product P3N-PIPO (26.3 kDa) is predicted to result from transcriptional slippage in the GA7T motif at nucleotide position 2918 within the P3 coding region and ending with a TAA stop codon at position 3147 (Olspert et al., 2015). NIa-Pro, the cysteine protease responsible for cleavage at the P3/6K1, 6K1/CI, CI/6K2, 6K2/VPg, VPg/NIa-Pro, NIa-Pro/NIb, NIb/CP junctions is predicted to recognize the consensus [DEC]-x-V-x-[YT]-[QE]/ [AGS].
Phylogenetic analysis of the putative polyprotein confirmed that PeMoV-pintoi is a member of the PeMoV clade while phylogenies constructed using the coherently evolving CP region showed PeMoV-pintoi to be most closely related to Australian isolate PeMoV-AU found to infect A. hypogea (Teycheney and Dietzgen, 1994). The PeMoV-pintoi/ PeMoV-AU group is part of a larger clade comprising sequences isolated in the United States and Iran, all infecting peanut and clearly separated from strains infecting soybean. PeMoV was first described in 1965 in the United States and since then it has been reported to infect peanut in Africa, Asia, Australia and South America (Kuhn 1965; Sreenivasulua and Demski, 1988; Soumya et al., 2014). PeMoV generally induces greenish mottle symptoms in peanuts and is also known to infect common bean and Cassia sp. (Kuhn, 1965; Lim et al., 2014). PeMoV is of quarantine significance, can be transmitted by seeds and in a non-persistent manner by aphids such as Aphis craccivora and Myzus persicae (Adams and Kuhn, 1977; Sreenivasulua and Demski, 1988).
The closest PepMov strain comes from Australia, where it is widely cultivated as forage in New South Wales and South-Eastern Queensland (Jones 1993; Bowman et al., 1998). Australia was a source of Arachis accessions used in the selection of the best cultivars suited for use in Colombia (Rincón et al., 1992); therefore, in spite of the large geographical distance between these two countries, the similarity between the Colombian and Australian PepMoV isolates comes as no surprise. In 1991, Morales and collaborators characterized the causal agent of foliar ring spot symptoms in A. pintoi to be serologically related to PeMoV and which could also cause systemic infections in other legumes such as common bean, cowpea, peanut and soybean (Morales et al., 1991). Unfortunately, monoclonal antibodies failed to confirm this suggestion. Our results confirm that PeMoV can naturally infect A. pintoi and, to our knowledge, this is first complete PeMoV genome sequence reported in a host different to A. hypogaea.
Polerovirus
Poleroviruses are members of the Luteoviridae family, which are characterized by isometric virions of about 25 to 30 nm in diameter that encapsidate a positive single stranded genomic RNA of 5.6 kb to 6.0 kb with a Vpg protein linked to the 5' end and lack of a poly-A tail (King et al., 2012). The assembly of the polerovirus present in the sample resulted in a contig of 5578 nt with an average sequence coverage of 724x, which was named TuYV-pintoi. Twenty seven polymorphic sites were found at positions 14, 161, 234, 434, 662, 778, 882, 888, 900, 1410, 1428, 1702, 1745, 2355, 2660, 2960, 3095, 4115, 4181, 4199, 4292, 4425, 4561, 4702, 4725, 5078 and 5226; the observed transition/transversion ratio was 3.5 (Figure 4). The best BLASTN hits were Turnip yellows virus isolates BWYV-FL1 (gb:X13063, 91%) and WA-1 (gb: JQ862472, 91%) identified in France (Veidt et al., 1988) and Australia (Wylie et al., 2013), respectively.
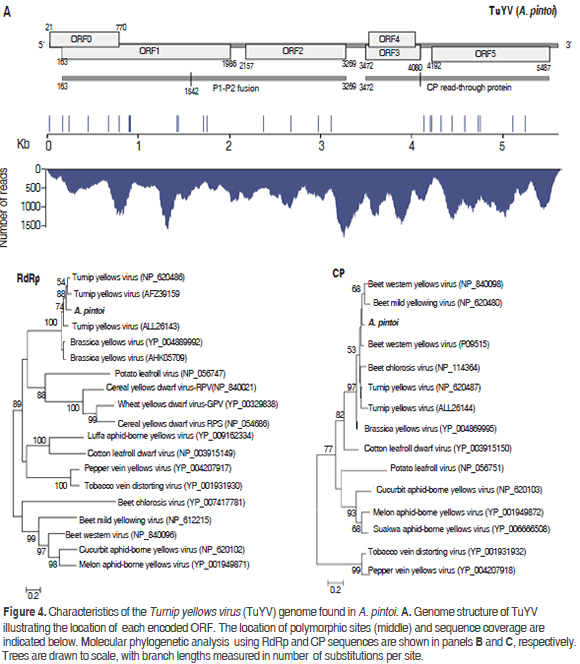
TuYV-pintoi contains six open reading frames ORF0-ORF5 at positions 21-770, 163-1986, 2157-3269, 3472-4080, 3503-4030 and 4192-5487, respectively, in agreement with the genomic structure of the genus Polerovirus (King et al., 2012). ORF0 is predicted to encode protein P0 (28.9 kDa) that functions as a suppressor of RNA silencing and is involved in symptom and host range determination (Pfeffer et al., 2002). ORF1 has a 607 nt overlap with ORF0 and codes for a 66.21 kDa protein (P1) containing an endopeptidase domain (pfam02122) at residues 206-408. This protein is believed to play a major role in the replication cycle by promoting the maturation of the genome-linked virion protein VPg (Nickel et al., 2008). ORF2 is the RNA-dependent RNA polymerase (RdRp, 41.40kDa); however, as observed in other poleroviruses, a P1-P2 fusion protein of 115.56 kDa resulting from a -1 frameshift at position 1542 within ORF1 is predicted (Xiang et al., 2011) (Figure 4). ORF3 encodes a CP protein of 22.44 kDa protein expected to be produced by leaky scanning (King et al., 2012). ORF4 codes for a 19.71 kDa protein with luteovirus VPg genome linked protein motif (pfam01659) at positions 45-148 (Rathjen et al., 1994); this ORF is completely embedded within ORF3. ORF5 is predicted to encode a 47.89 kDa protein product; however, a P3-P5 read-through protein has been observed as a result of suppression at the amber stop codon of ORF3 (Stevens et al., 2005). With respect to the polymorphic sites in the consensus sequence, two resulted in amino acid substitutions in P0 (N47K and Y72H), six in the P1-P2 fusion (A91V, T167M, V206I, N462K, N414S, I732V) and six in the CP read-through (P215L, A237V, Q243L, S274F, N364Y, Y421H, I418M and L536P).
Phylogenetic analysis using the RdRp protein confirmed TuYV-pintoi to be closely related to Turnip yellows virus and, more distantly to Brassica yellows virus. With respect to the CP analysis, TuYV-pintoi clustered within a group comprising Beet western yellows virus, Beet mild yellowing virus, Beet western yellows virus and Beet chlorosis virus. This clade also includes TuYV and Brassica yellows virus with a bootstrap of 97%. In the RdRp phylogenetic tree these viruses were separated into a different group. Interspecific and intraspecific phylogenetic studies suggested that beet-infecting polerovirus arose by recombination between a CABY-like ancestor contributing to ORFs 0, 1, 2 and a beet polerovirus parent that provided the 3'ORFs (Hauser et al., 2002; Beuve et al., 2008; Zhou et al., 2011) thus explaining the conflicting results between CP and RdRp trees (Figure 4).
TuYV was originally identified in the United States in the late 1950s and has been reported to infect a wide range of hosts such as Brassica napus (oilseed rape), peanut, Gomphrena globosa (globe amaranth), Crambe abyssinica, Trifolium subterraneum (subterranean clover), Montia perfoliata (Indian lettuce), Lactuca sativa (lettuce), Capsella bursa-pastoris (Shepherd´s purse), Pisum sativum (common pea), Glycine max (soybean), Lens culinaris (lentil), Senecio vulgaris (common groundsel) and Spinacia oleracea (spinach) (King et al., 2012). Symptoms may include systemic leaf reddening, leaf yellowing and stunting (King et al., 2012; Lim et al., 2014). TuYV can be carried and transmitted in a persistent manner by aphids (Homoptera, Aphididae) such as Macrosiphum euphorbiae, Aphis fabae, A. gossypii, Myzus ascalonicus and M. persicae, which seems to be the most efficient (Stevens et al., 2005). The transmission of TuYV-pintoi by aphids should to be addressed in future studies. The genome presented here is the first complete genome of a putative polerovirus naturally infecting A. pintoi.
Alphaflexiviridae
The analysis of reads evidenced the existence of viral sequences related to the family Alphaflexiviridae with an overall nucleotide percent identity of 85% to currently accepted genera by the ICTV (Figure 5). This family comprises viruses with genomes consisting of a single positive linear ssRNA of about 5.9-9.0 kb, capped with m7G at the 5' end and containing a 3' poly(A) tract (Martelli et al., 2007). Alphaflexiviridae typically encode 5 to 6 ORFs which include a RNA polymerase, triple gene block (TGB) proteins involved in cell-to-cell movement, a coat protein and, in some genera, a sixth ORF containing a zinc finger motif with nucleic acid binding activity. Currently, the Alphaflexiviridae family is divided into six Genera: Allexivirus, Botrexvirus, Lolavirus, Mandarivirus, Potexvirus and Sclerodarnavirus (King et al., 2012).
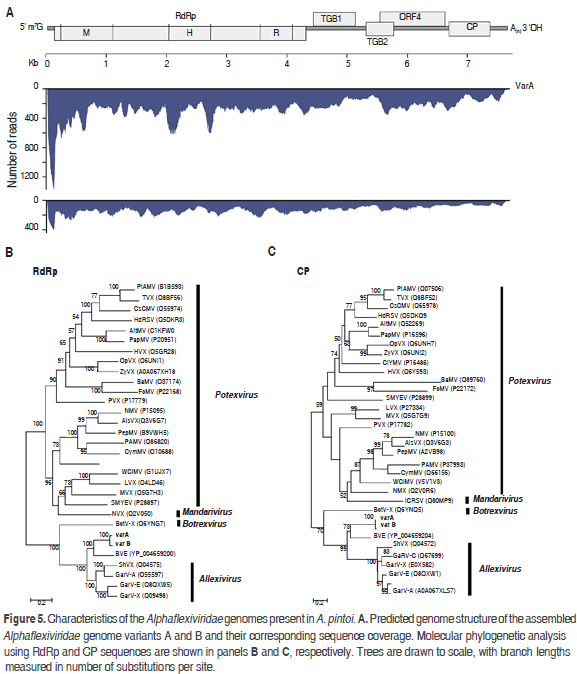
De novo assembly resulted in two distinct contigs of 7561(135x) and 7538 (260x) nt, that share 85% of nucleotide identity and representing two variants (A and B). The most similar viruses in the NCBI database are Blackberry virus E (BVE) and Garlic virus A with 54 and 50 percent nucleotide sequence identity, respectively. BVE belongs to an unassigned genus within the family Alphaflexiviridae related to allexiviruses but lacks a 3'end-proximal ORF (Sabanadzovic et al., 2011). Garlic virus A (GarV-A), on the other hand, is a member of the genus Allexivirus which also includes ShVX, Garlic mite-borne filamentous virus (GarMbFV), Garlic virus B (GarV-B), Garlic virus C (GarV-C), Garlic virus D (GarV-D), Garlic virus E (GarV-E) and, Garlic virus X (GarV-X). The assembled genomes contain five ORFs identified at positions 129-4307 (ORF1), 4396-5109 (ORF2), 5087-5407 (ORF3), 5509-6597 (ORF4) and 6650-7345 (ORF5). ORF1 encodes a putative RNA-dependent RNA polymerase of 1392 residues (157.61kDa) with characteristic methyltransferase (pf01660, 40-343), helicase (PF01443, 636-869) and replicase (PF00978, 991-1338) domains. ORF2 codes for a 26.46 kDa protein and contains an ATPase (pfam13191) and viral RNA helicase (pfam01443) domains at positions 12-138 and 26-227, respectively. ORF2 and ORF3 are homologous to the triple gene block proteins TGBp1 and TGBp2. ORF4 encodes a 40.84 kDa protein homologous to the 40kDa protein of Alphaflexiviridae. ORF5 encodes a 25.52 kDa protein homologous to the viral coat protein of Alphaflexiviridae. No sequences homologous to ORF6 of allexiviruses were identified in the assembled genomes. The same number of ORFs was found in Variant B.
Phylogenetic analysis using the RdRp and CP proteins revealed that variants A and B are a sister group of BVE and are closely related to the genus Allexivirus (Figure 5); we propose the names Arachis pintoi virus A and Arachis pintoi virus B for these isolates. Our data indicates that variants A and B probably belong to a new genus within the family Alphaflexiviridae; however future work should confirm the existence of this proposed new genus using specific primers and by satisfying Koch´s Postulates.
CONCLUSIONS
Using Next-Generation sequencing of the A. pintoi transcriptome from a bulk sample of leaves, three different virus species from the Potyviridae, Luteoviridae and Alphaflexiviridae families were identified. PeMoV and TuYV have been previously reported infecting peanut; in contrast, the Alphaflexiviridae member probably corresponds to a new virus distantly related to the genus Allexivirus and which would be a first report of this family in A. pintoi.
The presence of at least three viruses infecting A. pintoi in Colombia suggests that it is important to initiate a germplasm clean-up program in the seed and propagation material used for this plant. Further studies should also address the transmission mechanisms of these viruses, their incidence across the country, as well as their host range.
ACKNOWLEDGMENTS
This work was supported by the Faculty of Sciences, Universidad Nacional de Colombia Sede Medellín (Código Hermes 32098 "Desarrollo de una plataforma bioinformática para la detección y el estudio del procesamiento postraduccional de Potyvirus". Convocatoria Apoyo a Grupos de Investigación - Facultad de Ciencias).
REFERENCES
Adams DB and Kuhn CW. 1977. Seed transmission of Peanut mottle virus in Peanuts. Phytopathology 67(9): 1126-1129. [ Links ]
Adams MJ, Antoniw JF and Beaudoin F. 2005. Overview and analysis of the polyprotein cleavage sites in the family Potyviridae. Molecular Plant Pathology 6(4): 471-87. doi: 10.1111/j.1364-3703. 2005.00296.x [ Links ]
Adams IP, Glover RH, Monger WA, Mumford R, Jackeviciene E, Navalinskiene M, Samuitiene M and Boonham N. 2009. Next-generation sequencing and metagenomic analysis: a universal diagnostic tool in plant virology. Molecular Plant Pathology 10(4): 537-545. doi: 10.1111/j.1364-3703.2009.00545.x [ Links ]
Beuve M, Stevens M, Liu HY, Wintermantel WM, Hauser S and Lemaire O. 2008. Biological and molecular characterization of an American sugar beet-infecting Beet western yellows virus isolate. Plant Disease 92(1): 51-60. doi: 10.1094/PDIS-92-1-0051 [ Links ]
Bowman AM, Wilson GPM and Gogel BJ. 1988. Evaluation of perennial peanuts (Arachis spp.) as forage on the New South Wales coast. Tropical Grasslands 32: 252-258. [ Links ]
Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32(5): 1792-1797. doi: 10.1093/nar/gkh340 [ Links ]
Gish W and States DJ. 1993. Identification of protein coding regions by database similarity search. Nature Genetics 3(3): 266-272. doi: 10.1038/ng0393-266 [ Links ]
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N and Regev A. 2011. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nature Biotechnology 29(7): 644-652. doi: 10.1038/nbt.1883 [ Links ]
Gutiérrez PA, Alzate JF and Marín M. 2014a. Genome sequence of a virus isolate from tamarillo (Solanum betaceum) in Colombia: evidence for a new potyvirus. Archives of Virology 160(2): 557-560. doi: 10.1007/s00705-014-2296-8 [ Links ]
Gutiérrez PA, Alzate JF and Marín M. 2014b. Caracterización del viroma de ARN de tejido radical de Solanum phureja mediante pirosecuenciación 454 GS-FLX. Bioagro 26(2): 89-98. [ Links ]
Gutiérrez PA, Alzate JF and Marín M. 2015. Complete genome sequence of an isolate of Potato virus X (PVX) infecting Cape gooseberry (Physalis peruviana) in Colombia. Virus Genes 50(3): 518-522. doi: 10.1007/s11262-015-1181-1 [ Links ]
Gutiérrez P, Mesa H and Marín M. 2016. Genome sequence of a divergent Colombian isolate of Potato virus V (PVV) infecting Solanum phureja. Acta Virologica 60(1):49-54. doi: 10.4149/av_2016_01_49 [ Links ]
Hasegawa M, Kishino H and Yano T. 1985. Dating the human-ape split by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution 22(2): 160-174. doi: 10.1007/BF02101694 [ Links ]
Hauser S, Stevens M, Beuve M and Lemaire O. 2002. Biological properties and molecular characterization of Beet chlorosis virus (BChV). Archives of Virology 147(4): 745-762. doi: 10.1007/s007050200023 [ Links ]
Jones RM. 1993. Persistence of Arachis pintoi cv. Amarillo on three soil types at Samford, South-Eastern Queensland. Tropical Grasslands 27:11-15. [ Links ]
King AMQ, Adams MJ, Carstens EB and Lefkowitz EJ. 2012. Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press, San Diego, EEUU. 1338 p. [ Links ]
Krapovickas A and Gregory WC. 1994. Taxonomía del género Arachis (Leguminosae). Bonplandia 8(1): 1-186. [ Links ]
Kreuze J, Koenig R, De Souza J, Vetten HJ, Muller G, Flores B, Ziebell H and Cuellar W. 2013. The complete genome sequences of a Peruvian and a Colombian isolate of Andean potato latent virus and partial sequences of further isolates suggest the existence of two distinct potato-infecting tymovirus species. Virus Research 173(2): 431-435. doi: 10.1016/j.virusres.2013.01.014 [ Links ]
Kuhn CW. 1965. Symptomatology, host range and effect on yield of seed transmitted peanut virus. Phytopathology 55(8): 880-884. [ Links ]
Langmead B, Trapnell C, Pop M and Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology 10(3): R25. doi: 10.1186/gb-2009-10-3-r25 [ Links ]
Lascano CE and Thomas D. 1988. Forage quality and animal selection of Arachis pintoi in association with tropical grasses in the eastern plains of Colombia. Grass and Forage Science 43(4): 433-439. doi: 10.1111/j.1365-2494.1988.tb01900.x [ Links ]
Lavia GI, Ortiz AM, Robledo G, Fernández A and Seijo G. 2011. Origin of triploid Arachis pintoi (Leguminosae) by autopolyploidy evidenced by FISH and meiotic behavior Annals of Botany 108(1): 103-111. doi: 10.1093/aob/mcr108 [ Links ]
Le SQ and Gascuel O. 2008. An improved general amino acid replacement matrix. Molecular Biology and Evolution 25(7): 1307-1320. doi: 10.1093/molbev/msn067 [ Links ]
Lim S, Lee YH, Igori D, Zhao F, Yoo RH, Lee SH, Baek IY and Moon JS. 2014. First Report of Peanut mottle virus infecting soybean in South Korea. Plant Disease 98(9): 1285. doi: 10.1094/PDIS-04-14-0356-PDN [ Links ]
Martelli GP, Adams MJ, Kreuze JF and Dolja VV. 2007. Family Flexiviridae: a case study in virion and genome plasticity. Annual Review of Phytopathology 45(1): 73-100. doi: 10.1146/annurev.phyto.45.062806.094401 [ Links ]
Morales FJ, Castano M, Velasco AC and Arroyave J. 1991. Natural infection of tropical forage legume species of Arachis and Stylosanthes by potyviruses related to Peanut mottle virus. Plant Disease 75(11): 1090-1093. doi: 10.1094/PD-75-1090 [ Links ]
Nickel H, Kawchuk L, Twyman RM, Zimmermann S, Junghans H, Winter S, Fischer R and Prüfer D. 2008. Plantibody-mediated inhibition of the Potato leafroll virus P1 protein reduces virus accumulation. Virus Research 136(1-2): 140-145. doi: 10.1016/j.virusres.2008.05.001 [ Links ]
Nischwitz C, Maas AL, Mullis RM, Culbreath AK and Gitaitis RD. 2007. First report of Peanut mottle virus in forage peanut (Arachis glabrata) in North America. Plant Disease 91(5): 632. doi: 10.1094/PDIS-91-5-0632A [ Links ]
Olspert A, Chung BY, Atkins JF, Carr JP and Firth AE. 2015. Transcriptional slippage in the positive-sense RNA virus family Potyviridae. EMBO Reports 16(8): 995-1004. doi: 10.15252/embr.201540509 [ Links ]
Palmieri DA, Bechara MD, Curi RA, Monteiro JP, Valente SES, Gimenes MA and Lopes CR. 2010. Genetic diversity analysis in the section Caulorrhizae (genus Arachis) using microsatellite markers. Genetics and Molecular Biology 33(1): 109-118. doi: 10.1590/S1415-47572010005000001 [ Links ]
Pfeffer S, Dunoyer P, Heim F, Richards KE, Jonard G and Ziegler-Graff V. 2002. P0 of Beet western yellows virus is a suppressor of posttranscriptional gene silencing. Journal of Virology 76(13): 6815-6824. doi: 10.1128/JVI.76.13.6815-6824.2002 [ Links ]
Posada D and Crandall KA. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14(9): 817-818. doi: 10.1093/bioinformatics/14.9.817 [ Links ]
Rathjen JP, Karageorgos LE, Habili N, Waterhouse PM and Symons RH. 1994. Soybean dwarf luteovirus contains the third variant genome type in the luteovirus group. Virology 198(2): 671-679. doi: doi: 10.1006/viro.1994.1079 [ Links ]
Rincón CA, Cuesta PA, Pérez R, Lascano CE and Ferguson J. 1992. Maní forrajero perenne (Arachis pintoi Krapovickas y Gregory). Una alternativa para ganaderos y agricultores. Boletín Técnico No. 219. ICA/CIAT, Palmira. 23 p. [ Links ]
Rincón A. 2001. Potencial productivo de ecotipos de Arachis pintoi en el Piedemonte de los Llanos Orientales de Colombia. Pasturas Tropicales 23: 19-24. [ Links ]
Roossinck MJ, Martin DP and Roumagnac P. 2015. Plant virus metagenomics: Advances in virus discovery. Phytopathology 105(6): 716-27. Doi: 10.1094/PHYTO-12-14-0356-RVW [ Links ]
Sabanadzovic S, Abou N and Tzanetakis IE. 2011. Blackberry virus E: an unusual flexivirus. Archives of Virology 156(9): 1665-1669. doi: 10.1007/s00705-011-1015-y [ Links ]
Soumya K, Yogita M, Prasanthi Y, Anitha K, Kavi-Kishor PB, Jain RK and Mandal B. 2014. Molecular characterization of Indian isolate of Peanut mottle virus and immunodiagnosis using bacterial expressed core capsid protein. Virus disease 25(3): 331-337. doi: 10.1007/s13337-014-0210-3. [ Links ]
Sreenivasulu P and Demski JW. 1988. Transmission of Peanut mottle and Peanut stripe viruses by Aphis craccivora and Myzus persicae. Plant Disease 72:722-723. doi: 10.1094/PD-72-0722 [ Links ]
Sreenivasulu P, Reddy CVS, Ramesh B and Kumar PL. 2008. Groundnut viruses. pp. 47-97. En: Rao GP, Khurana SMP and Lenardon SL. Characterization, diagnosis & management of plant viruses. Volume 1: industrial crops. Studium Press, India. [ Links ]
Stevens M, Freeman B, Liu HY, Herrbach E and Lemaire O. 2005. Beet poleroviruses: close friends or distant relatives? Molecular Plant Pathology. 6: 1-9. doi: 10.1111/j.1364-3703.2004.00258.x [ Links ]
Tamura K, Stecher G, Peterson D, Filipski A and Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular Biology and Evolution 30(12): 2725-2729. doi: 10.1093/molbev/mst197. [ Links ]
Teycheney PY and Dietzgen RG. 1994. Cloning and sequence analysis of the coat protein genes of an Australian strain of Peanut mottle and an Indonesian 'blotch' strain of peanut stripe potyviruses. Virus Research 31(2): 235-244. doi: 10.1016/0168-1702(94)90006-X [ Links ]
Valls JFM. 1996. Variability in the genus Arachis and potential forage uses. pp 15-27. En: Springer TL and Pittman RN (eds). Identifying germplasm for succesful forage legume-grass interations. Proceedings of a Symposium of the Crop Science Society of America. USDA, Washington. [ Links ]
Veidt I, Lot H, Leiser M, Scheidecker D, Guilley H, Richards K and Jonard G. 1988. Nucleotide sequence of Beet western yellows virus RNA. Nucleic Acids Research 16(21): 9917-9932. doi: 10.1093/nar/16.21.9917 [ Links ]
Villamil-Garzón A, Cuellar WJ and Guzmán-Barney M. 2014. Natural co-infection of Solanum tuberosum crops by the Potato yellow vein virus and potyvirus in Colombia. Agronomía Colombiana 32(2): 213-223. doi: 10.15446/agron.colomb.v32n2.43968 [ Links ]
Wu Q, Ding S, Zhang Y and Zhu S. 2015. Identification of viruses and viroids by Next-Generation Sequencing and homology dependent and homology independent algorithms. Annual Review of Phytopathology 53: 1-20. doi: 10.1146/annurev-phyto-080614-120030 [ Links ]
Wylie SJ, Li H, Dixon KW, Richards H and Jones MG. 2013. Exotic and indigenous viruses infect wild populations and captive collections of temperate terrestrial orchids (Diuris species) in Australia. Virus Research 2013 171(1): 22-32. doi: 10.1016/j.virusres.2012.10.003 [ Links ]
Xiang HY, Dong SW, Shang QX, Zhou CJ, Li DW, Yu JL and Han CG. 2011. Molecular characterization of two genotypes of a new polerovirus infecting brassicas in China. Archives of Virology 2011 156(12): 2251-2255. doi: 10.1007/s00705-011-1091-z [ Links ]
Zhou CJ, Xiang HY, Zhuo T, Li DW, Yu JL and Han CG. 2011. A novel strain of Beet western yellows virus infecting sugar beet with two distinct genotypes differing in the 5'-terminal half of genome. Virus Genes 42(1):141-149. doi: 10.1007/s11262-010-0553-9 [ Links ]

