










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActualidades Biológicas
Print version ISSN 0304-3584
Actu Biol vol.34 no.96 Medellín Jan./June 2012
ARTÍCULOS DE INVESTIGACIÓN
DIVERSIDAD GENÉTICA EN EL BANCO DE GERMOPLASMA DE MORERA [MORUS SPP. (ROSALES: MORACEAE)] DE LA GRANJA EXPERIMENTAL ''EL PILAMO'', UNIVERSIDAD TECNOLÓGICA DE PEREIRA, PEREIRA (RISARALDA), COLOMBIA
GENETIC DIVERSITY IN THE GERMPLASM BANK OF MULBERRY [MORUS SPP. (ROSALES: MORACEAE)] BELONGING TO THE ''EL PILAMO'' EXPERIMENTAL FARM, UNIVERSIDAD TECNOLÓGICA DE PEREIRA, PEREIRA (RISARALDA), COLOMBIA
Duverney Gaviria-Arias1,2; Enrique Aguilar-Fernández1,3; Heidy Navia-Morocho1,4; Álvaro Alegría-Soto1,5
1 Centro de Biología Molecular y Biotecnología, Facultad de Ciencias de la Salud, Universidad Tecnológica de Pereira. Vereda ''La Julita'', Pereira (Risaralda), Colombia.
Correos electrónicos: 2 duverney.gaviria@gmail.com; 3 eaf578@yahoo.com; 4 heidycatalina@gmail.com; 5 cenbiotep@utp.edu.co.
Recibido: octubre 2011. Aceptado: abril 2012.
Resumen
Usando los polimorfismos de longitud de fragmentos amplificados (AFLPs) se analizó la diversidad genética de 31 accesiones de morera, Morus spp. (Moraceae) del banco de germoplasma de la granja experimental ''El Pílamo'' de la Universidad Tecnológica de Pereira (Colombia). Se utilizaron 5 combinaciones de iniciadores AFLPs que generaron 152 bandas polimórficas con una correlación promedio del 20% y permitieron analizar el 64,12% de la diversidad total de estas accesiones. Se evaluaron la diversidad genética, las relaciones existentes entre las accesiones y el grado de estructuración. Los resultados de las combinaciones de iniciadores mostraron valores de heterocigosidad promedio de 0,2332 y diversidad genética de 0,2302. El análisis de conglomerados mostró que cada una de las 31 accesiones tiene diferente genotipo. Cinco grupos definidos fueron así establecidos. El análisis de AMOVA permitió establecer que el 77% de la diversidad estaba dentro de la población y el 23% entre los grupos definidos molecularmente. Los grupos mostraron un índice FST de 0,235 y un valor de flujo de genes de 1,1. Se concluye que los grupos establecidos molecularmente mostraron valores elevados a nivel de distancia genética y loci polimórficos, especialmente las poblaciones 1 y 5. Se concluye que el genoma de morera presenta un alto grado de complejidad como se demuestra de acuerdo con la colección estudiada.
Palabras clave: AFLPs, caracterización molecular, estructura poblacional, Morus spp., Colombia.
Abstract
TUsing amplified fragment length polymorphisms (AFLPs), the genetic diversity of 31 accessions of the mulberry, Morus spp. (Moraceae) from the germplasm collection of the experimental farm ''El Pílamo'' (Universidad Tecnológica de Pereira, Colombia) were analyzed. Five primer combinations generated 152 polymorphic AFLP bands with an average correlation of 20% and allowed analysis of 64.12% of the total diversity present in these accessions. Genetic diversity, relationships among the accessions and the degree of population structure were evaluated. The primer combinations showed mean heterozygosity and genetic diversity values of 0.2332 and 0.2302, respectively. Cluster analysis showed that each of the 31 accessions has a different molecular genotype. Five groups were recognized. AMOVA analysis revealed that 77% of the diversity was within populations and 23% between the recognized molecular groups. The groups exhibited an FST value of 0.235 and a gene flow value of 1.1. The molecularly established groups showed elevated levels of both genetic distance and polymorphic loci, especially populations 1 and 5. We conclude that mulberry has high genomic complexity, as evidenced in the collection investigated here.
Key words: AFLPs, molecular characterization, population structure, Morus spp.
INTRODUCCIÓN
El género Morus L. (Moraceae), es uno de grupos taxonómicos más interesantes por su gran variabilidad genética y su importancia comercial (Vijayan et al. 2011). La familia Moraceae se subdivide en 4 subfamilias, 55 géneros y cerca de 950 especies, cada una con miles de variedades (Fotadar y Dandin 1998, Hirano 1982 y Vijayan et al. 2011). La morera se distribuye ampliamente en el mundo, desde regiones tropicales hasta regiones subarticas (Awasthi et al. 2004, Sharma et al. 2000). Sus hojas son utilizadas en bebidas y sus frutos son consumidos frescos o en forma de jugos o conservas (Kalpana et al. 2012). Adicionalmente la morera se siembra para prevenir la erosión en suelos que están sometidos a la acción del agua o del viento y es utilizada como forraje para diversas especies de ganado por su alto valor nutritivo, expresado en sus elevados niveles de proteína (Sánchez 2000, Vijayan et al. 2011). Recientemente, se ha prestado mucha atención al uso de productos naturales con propiedades cosmeceúticas y la familia Moracea es una de las más interesantes en este sentido, ya que a partir de las especies de Morus se ha podido lograr la extracción de productos con actividad hipoglicemiante, azúcares nitrogenados, hormonas vegetales, saponinas, moracetinas y diversos tipos de metabolitos secundarios de tipo polifenólico como rutinina, isoquercetina y astragalina (Datta 2000).
A pesar de la dificultad en la caracterización de morera, se pueden diferenciar tres especies principales, 1) Morus alba L. o morera blanca distribuida principalmente en la región norte y sureste de Asia y, centro y noreste de China; 2) Morus nigra L. o morera negra cuyo origen se encuentra en el oriente de Asia y en el continente Europeo y; 3) Morus rubra L. o la morera roja nativa de las Américas. Cada una de ellas presenta características de adaptación propias de sus respectivos orígenes. Sin embargo, M. alba por su gran resistencia y adaptabilidad a condiciones de sequía y suelos pobres, es la de mayor distribución en el mundo aún en áreas urbanas (Sánchez 2000, Sharma et al. 2000, Vijayan et al. 2011).
El auge de la sericultura a nivel mundial ha llevado a que muchos de los cultivares actuales hayan sufrido procesos de naturalización al ser transportadas de su sitio de origen a regiones donde generalmente no se desarrollaban (organismos alóctonos), otorgándoles nuevos nombres y provocando en muchos casos confusiones en los procesos de evaluación taxonómica (Sharma et al. 2000). La clasificación del germoplasma de morera ha sido llevada a cabo tradicionalmente por métodos de sistemática convencional estudiando la morfología de la hoja, la longitud de los estilos en las flores femeninas, la forma de los idioblastos, la forma y número de los cistolitos y el color de la fruta (Katsumata 1971). Sin embargo, dada la gran diversidad dentro del género y lo influenciables que son este tipo de caracteres por el entorno ambiental, las clasificaciones basadas en estos caracteres no son precisas (Cappellozza et al. 1995). Por estas razones se han realizado algunas aproximaciones a nivel molecular sobre la diversidad del género Morus, estas han permitido el estudio de las identidades genéticas y de las relaciones entre las plantas de morera. Las aproximaciones han sido realizadas con marcadores basados en ADN como los AFLP (Sharma et al. 2000; Wang y Yu 2001, Yang et al. 2003), DAMD (Bhattacharya et al. 2001), ISSR (Vijayan y Chatterjee 2003, Zhao et al. 2006, Zietkiewicz et al. 1994), RAPD (Awasthi et al. 2004, Esha y Shirish 2001, Xiang et al. 1995, Zhao y Pan 2004, Zhao et al. 2000) y SSR (Zhao et al. 2005). Los cuales son altamente heredables, fácilmente observables y menos influenciados por el ambiente.
En Colombia, el desarrollo de la industria de la sericultura ha sido promovido en varias regiones como una alternativa al trabajo de los agricultores. Debido a este proceso la granja experimental ''El Pilamo'' posee un banco de germoplasma con 31 accesiones del género Morus, las cuales en la actualidad no han sido evaluadas por sus características taxonómicas, medicinales y para la obtención de metabolitos secundarios. El objetivo de este estudio fue evaluar la diversidad genética y tipos de relaciones entre las diferentes accesiones mantenidas en la granja experimental ''El Pilamo'', mediante el uso de marcadores moleculares AFLPs, estableciendo de esta manera una línea base para iniciar los estudios tendientes a aumentar el conocimiento de este importante recurso vegetal y aprovechar al máximo sus características.
MATERIALES Y MÉTODOS
Material vegetal y extracción de ADN. El estudio se desarrolló sobre 31 accesiones de morera mantenidas en la granja experimental ''El Pilamo'' de la Universidad Tecnológica de Pereira, vereda ''La Honda, aproximadamente a 16 km del área urbana del municipio de Pereira (Risaralda), Colombia, a una altura de 1.100 m con coordenadas geográficas: 410607 N, 538911 O, precipitación anual promedio de 3.000 mm y temperatura promedio de 23 °C. La colección consta de 11 variedades de M. alba, 1 variedad de M. indica, 1 variedad de M. formosiensis, 1 variedad de M. acidoasa y 17 variedades no identificadas de morera Morus spp. Todas estas variedades fueron adquiridas en diversas partes del mundo por parte del Centro de Desarrollo Tecnológico de la Sericultura (CDTS), antigua granja experimental ''El Pilamo'', como parte de su misión institucional.
La extracción del material genético se llevó a cabo siguiendo el método propuesto por Doyle y Doyle (1990), utilizando hojas (1 g) maceradas en nitrógeno líquido. La evaluación de las características del material genético extraído en términos de cantidad e integridad (no degradación) se llevó a cabo mediante electroforesis en geles de agarosa al 1% teñido con bromuro de etidio. La cuantificación del ADN se realizó utilizando un espectrofotómetro UV/VIS 1601 SHIMADZU con un resultado promedio de 500 ng/μl por muestra. El registro permanente de los datos se realizó usando una cámara digital Nikon D-100.
Polimorfismo en la longitud de los fragmentos amplificados. Las reacciones de AFLP se llevaron a cabo usando el estuche comercial ''AFLP® system I'' de la casa comercial Invitrogen, siguiendo las recomendaciones del fabricante (Vos et al. 1995). Se usaron 300 ng de ADN de cada una de las accesiones, los cuales fueron digeridos usando la combinación de enzimas EcoRI y MseI suministradas. Posteriormente, estos digeridos fueron ligados a adaptadores para cada una de las enzimas usadas y se llevó a cabo una pre-amplificación. Este producto fue diluido en Tris-HCl 10mM y EDTA 1mM (TE) en una proporción de 1:20 y usado en las reacciones siguientes de amplificación selectiva. En total fueron evaluadas 5 combinaciones de iniciadores que mostraron mayor polimorfismo de un grupo de 10 combinaciones.
Evaluación del resultado de amplificación selectiva. La separación de los fragmentos de ADN se realizó en gel de poliacrilamida desnaturalizante al 6% (19:1) y 7M de urea. Se tomaron 5μl del producto de PCR y se le agregó 5μl de amortiguador de carga (1M formamida, 20 mM EDTA, 0,05% xilene cianol, 0,05% azul de bromofenol). Los fragmentos fueron separados durante 90 min a 90 W. La visualización de los fragmentos de ADN se realizó mediante tinción con plata utilizando el estuche comercial Silver sequenceTm DNA sequencing system (Promega), siguiendo las recomendaciones del fabricante. Como marcador de peso molecular se usó la escalera de ADN 30-330 pb de la casa comercial Invitrogen.
Lectura de datos. La lectura de la información para cada par de iniciadores se realizó de manera manual señalando las bandas intensas y claramente definidas. El rango de lectura se estableció desde las 50 pb hasta 300pb, dado que en esta región se observaron bandas bien definidas y claramente analizables. Cada banda de ADN fue tomada como un locus genético, asumiendo además que los alelos marcadores de diferentes loci no comigran y que cada locus puede ser tratado como un sistema de dos alelos (Lynch y Milligan 1994). De esta manera se construyeron matrices de datos con 1 para la presencia y 0 para la ausencia de cada uno de los loci.
Análisis de datos. Las matrices obtenidas por cada una de las combinaciones fueron inicialmente evaluadas con el fin de determinar si proporcionaban o no la misma información y si el número de loci polimórficos de cada una de ellas había sido suficiente para analizar la diversidad presente en las accesiones. Esto se llevó a cabo con una prueba de correlación de matrices de Mantel (1967). El análisis de los datos se llevó a cabo preliminarmente utilizando solo un nivel jerárquico, agrupando a las 31 accesiones evaluadas en una sola población. A partir de la matriz binaria se calcularon los siguientes parámetros estadísticos: heterocigosidad, frecuencia alélica y porcentaje de loci polimórficos asumiendo para el análisis que las frecuencias alélicas en la población están en equilibrio de Hardy-Weinberg. Para esto se usaron los programas POPGEN 3.2 (Yeh et al. 1997) y GENALEX 6 (Peakall 2006). Se efectuó un análisis de los conglomerados utilizando los programas TFPGA (Miller 1997), NTSYS-pc versión 2.11 (Rohlf 2002) y POPGEN 3.2. Para esto se usaron dos estrategias de agrupamiento, el método de los pares de grupos no ponderados con media aritmética (UPGMA; Sneath y Sokal 1973) y un análisis de componentes principales utilizando los datos de distancia genética de Nei (1978). Los resultados de conglomerados sugirieron la conformación de 5 grupos que para el análisis estadístico se tomaron como poblaciones, aunque no en el sentido biológico. La conformación de los grupos no mostró asociación con las variedades presentes ya que a lo largo de los grupos establecidos se encontraron variedades de M. alba. La repetitividad de los agrupamientos fue evaluada con el uso de la estrategia Bootstrap en el programa TFPGA (Miller 1997) utilizando 5.000 iteraciones. Adicionalmente, la significancia fue evaluada mediante análisis de diferenciación poblacional, llevados a cabo con el uso de un AMOVA (Excoffier 1995), con el paquete estadístico GENALEX 6. Antes de realizar cada uno de los análisis, el supuesto de homocedasticidad fue revisado, mediante una prueba de Bartlett (Sokal y Rohlf 2012). Con el fin de comprobar la homocedasticidad, se determinaron los parámetros de variación genética dentro y entre poblaciones Fst en el programa POPGEN 3.2.
RESULTADOS Y DISCUSIÓN
El análisis arrojó un total de 246 bandas para las cinco combinaciones analizadas (tabla 1) con un porcentaje de loci polimórficos distribuidos entre 59,37-82,81%. Sin embargo, este total de bandas fue corregido con el fin de eliminar aquellos marcadores identificados como monomórficos. Dicho ajuste se realizó mediante la corrección para el manejo de marcadores dominantes establecida por Lynch y Milligan (1994). Realizada esta corrección se obtuvieron 152 bandas polimórficas sobre las cuales se llevaron a cabo los análisis descritos (tabla 1).
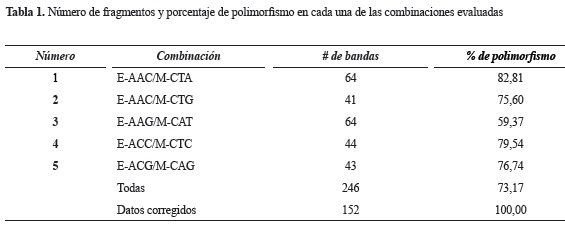
Con el fin de evaluar el grado de información suministrado por cada una de las combinaciones, se llevó a cabo una prueba de Mantel (1967). El grado de correlación entre las diferentes combinaciones mostró valores distribuidos desde un 5% aproximadamente hasta un 34% (tabla 2). La evaluación de la correlación de cada una de las combinaciones con la matriz correspondiente a la suma de todas las combinaciones, mostró valores entre 44 y 71%, siendo la matriz con mayor correlación la correspondiente a la combinación 3 (tabla 2); lo que parecería indicar que esta combinación evalúa regiones genómicas compartidas entre las diferentes accesiones.
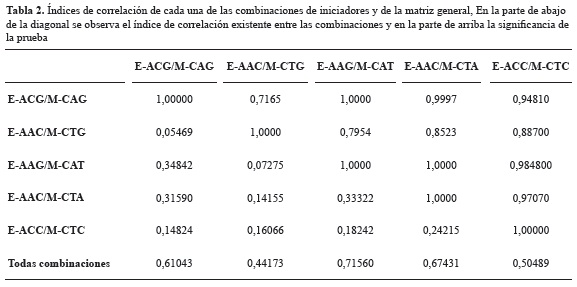
Adicionalmente y con el fin de determinar si las cinco combinaciones fueron suficientes para tener una adecuada evaluación de la diversidad genética, se correlacionaron los valores correspondientes al número de loci y la diversidad, identificándose que existía una correlación directa y positiva. Se derivó la siguiente ecuación, en donde ''y'' representa la diversidad genética y ''x'' el número de loci considerados:
y = 0,0034x
Se estableció que el 64,12% de la variación en la diversidad genética estimada es explicada por el número de loci analizados (r² = 0,6412). El análisis conjunto del grado de polimorfismo, los índices de correlación entre las diferentes matrices de datos, la correlación entre el número de loci y la diversidad estudiada muestran que en las accesiones analizadas existe un amplio conjunto de loci diferentes. Los bajos índices de correlación muestran que las combinaciones usadas podrían estar evaluando diferentes porciones en el genoma de morera o que existen niveles de ploidía diferentes entre las accesiones (Thome et al. 1996). El bajo índice de correlación entre la diversidad y el número de loci indicó que las combinaciones usadas no se encuentran evaluando ampliamente la diversidad presente en el genoma de morera y no fueron suficientemente informativas siendo necesario llevar a cabo la evaluación de un mayor número de combinaciones. Los resultados son comparables a los encontrados en otros trabajos en morera en términos de número de bandas analizadas y porcentajes de loci polimórficos (Kalpana et al. 2012, Vijayan et al. 2009, Zhao y Pan 2004). Los análisis de resultados para cada una de las combinaciones no fueron concordantes, demostrando esto la complejidad a nivel genómico que existe en estas plantas. Estos resultados están de acuerdo con los reportados por Bhattacharya y Ranade (2001), quienes con el uso de RAPDs y DAMD evaluaron la diversidad en morera encontrando conglomerados diferentes en cada técnica. Esta complejidad genómica en morera se reconoce de igual manera por la presencia de diferentes niveles de ploidía que en morera puede llegar hasta valores de 2 n = 22 X = 308 (docosaploide) (Vijayan et al. 2011, Yamanouchi et al. 2010).
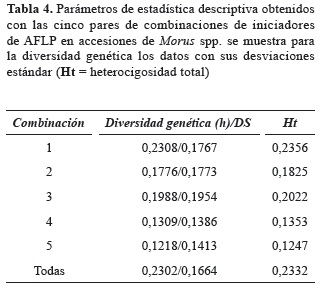
El análisis de estadística descriptiva en la población de morera permitió identificar que la diversidad genética fue de 0,2302 con una desviación estándar de 0,1664 calculada según Nei (1973) y el valor de Ht (Heterocigosidad total) fue de 0,2332 para la población completa (tabla 3), estos valores son más altos que los reportados en otros estudios (Zhao et al. 2006). Estos valores muestran que tanto la diversidad genética de la población como la riqueza de loci es alta, evidenciando la complejidad a nivel genómico en las accesiones de morera. Se identificó que la combinación que mayor diversidad genética y heterocigosidad mostró fue la combinación número 1, la cual presento adicionalmente el mayor nivel de polimorfismo (tabla 4).
El análisis de conglomerados evidenció que cada una de las variedades presentó genotipos diferentes por lo que cada una de ellas se localizó en hojas diferentes del árbol (figura 1), con una distancia genética distribuida entre 0,04 y 0,95, con un promedio de 0,495. Este valor indica que las diferentes accesiones de morera evaluadas comparten al menos 50% de sus loci, este valor es confirmado con el resultado del ajuste realizado al número de loci polimórficos, el cual bajó de 246 loci a 152 loci al eliminar aquellos que presentaron más de un 95% de monomorfismo (Lynch y Milligan 1994). La evaluación de conglomerados permitió inferir la conformación de 5 grupos (figuras 1 y 2) en los cuales la diversidad de M. alba, se mostró a los largo de todas las ramas (Zhao et al. 2005), esta observación parece confirmar la facilidad con la cual morera es capaz de establecer cruces interespecíficos lo que ha dificultado su conservación (Shabir et al. 2010). Dada la falta de información de pasaporte relacionada con estas accesiones que pudiese ser usada para la complementación de los análisis, se tomó solamente la información correspondiente a la evaluación molecular de este trabajo. En el análisis de conglomerados se observa que los grupos con mayor nivel de distancia genética fueron los correspondientes a los grupos 1 y 5, mientras que los grupos restantes revelan distribuciones más cerradas lo que indica que comparativamente presentan un menor nivel de diversidad.
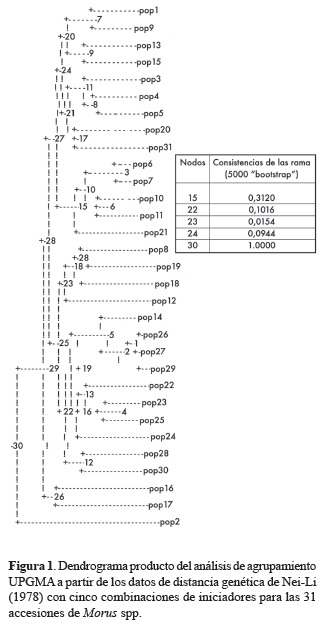
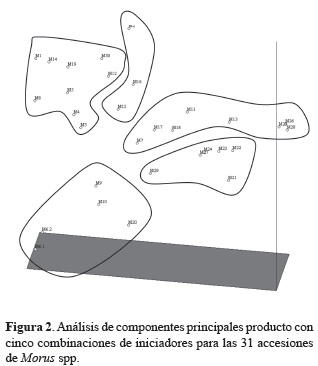
Con el fin de identificar si los conglomerados establecidos con el uso de los criterios anteriormente descritos eran sólidos o correspondían a artefactos del análisis, se realizó una evaluación de estructuración poblacional con el uso de AMOVA (tabla 4). Se identificó que el 77% de la diversidad encontrada era explicada por la diversidad dentro de la población y el 23% era explicado por la diversidad entre los grupos definidos. Los resultados de la AMOVA al igual que el valor de FST confirmaron que cada uno de los bloques sugeridos era significativo (Wright 1931). En todos los casos mostraron que existía un alto nivel de estructuración poblacional, con un nivel de flujo de genes bajo (Nm = 1,1). Estos valores en conjunto con los resultados anteriormente descritos muestran la gran complejidad presente en el genoma de morera ya que aún como se observa en el grupo 5, solamente tres individuos son suficientes para conformar un grupo sólido lo que hace referencia a la gran cantidad de información que aún falta por evaluar en estas accesiones.
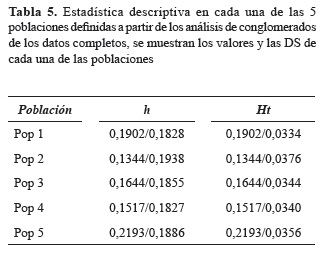
Dado que los análisis de diferenciación poblacional mostraron que los grupos conformados a partir del análisis de conglomerados fueron estadísticamente significativos, se llevó a cabo una evaluación de parámetros de estadística descriptiva para cada una de las 5 poblaciones establecidas (tabla 5). Los resultados muestran que las poblaciones con mayores niveles de distancia genética (h) y de polimorfismo a nivel de loci son las poblaciones 1 y 5, sin embargo, las otras poblaciones muestran valores significativamente elevados.
CONCLUSIONES
La evaluación de la diversidad genética existente en las accesiones de morera con el uso de los 152 marcadores de tipo AFLP resultó ser insuficiente, ya que solamente permitió identificar el 64% de la diversidad genética existente. Sin embargo, estos resultados no pudieron ser comparados con otros estudios porque en ninguno de ellos, fueron calculados estos valores. Los resultados fueron comparables en términos de número de loci totales y polimórficos y mejores que los registrados de diversidad genética y heterocigosidad en morera. Se hace necesario, por lo tanto, aumentar el número de loci evaluados. Los bajos valores de los coeficientes de correlación están informando la gran complejidad a nivel genómico presente en las 31 accesiones evaluadas pertenecientes al género Morus, ya que los resultados arrojados por cada una de las combinaciones, es decir la región del genoma que cada una de ellas está analizando es muy diferente. La única combinación que parece hacer un barrido general del genoma de morera es la combinación 3, ya que es la que presenta coeficientes de correlación más alto. La falta de información existente de las características biológicas de las accesiones de morera mantenidas en la granja experimental ''El Pilamo'' ha dificultado correlacionar los agrupamientos obtenidos con características específicas, sin embargo, al parecer y con la ayuda de los registros históricos de la colección, la diversidad de esta gira en torno a M. alba. Es evidente a partir de los resultados que todas las accesiones comparten entre sí más de 50% de sus loci lo que indica el alto grado de introgresión genética entre ellas. Sin embargo, los valores elevados en los aspectos de estadística descriptiva y diferenciación poblacional son igualmente sugerentes de su gran complejidad y por lo tanto de las amplias posibilidades que se podrían presentar al estudio de estas accesiones tanto con fines de mejoramiento genético como con la posibilidad de identificación de moléculas con actividad biológica importante.
REFERENCIAS
1. Awasthi AK, Nagaraja GM, Naik GV, Kanginakudru S, Thangavelu K, Nagaraju J. 2004. Genetic diversity and relationships in mulberry (genus Morus) as revealed by RAPD and ISSR marker assays. BMC Genetics, 5: 1-8. [ Links ]
2. Bhattacharya E, Ranade SA. 2001. Molecular distinction amongst varieties of mulberry using RAPD and DAMD profiles. BMC Plant Biology, 1 (3): 1-8. [ Links ]
3. Cappellozza L, Coradazzi AT, Tornadore N. 1995. Studies on the phenotypic variability of seven cultivars of Morus alba L and three of Morus multicaulis P. (Moraceae) - Part I. Sericologia, 35 (2): 257-270. [ Links ]
4. Datta RK [Internet]. 2000. Mulberry cultivation and utilization in India. FAO electronic conference on mulberry for animal production (Morus L). Fecha de acceso: 23 de enero de 2012. Disponible en: http://www.fao.org/DOCREP/005/X9895E/x9895e04. htm#TopOfPage. [ Links ]
5. Doyle JJ, Doyle JL. 1990. Isolation of plant DNA from fresh tissue. Focus, 12 (1): 13-15. [ Links ]
6. Excoffier L. 1995. Analysis of molecular variance (AMOVA) version 1.55. Geneva (Italia): LGB, University of Geneva. [ Links ]
7. Fotadar RK, Dandin SB. 1998. Genetic divergence in mulberry. Sericologia, 38 (1): 115-125. [ Links ]
8. Hirano H. 1982. Varietal differences of leaf protein profiles in mulberry. Phytochemistry, 21: 1513-1518. [ Links ]
9. Kalpana D, Hyuk Choi S, Ki Choi T, Senthil K, Soo Lee Y. 2012. Assessment of genetic diversity among varieties of mulberry using RAPD and ISSR fingerprinting. Scientia Horticulturae, 134: 79-87. [ Links ]
10. Katsumata F. 1971. Shape of idioblast in mulberry leaves with special reference to the classification of mulberry trees. Journal of Sericulture Science of Japan, 40 (4): 312-322. [ Links ]
11. Lynch M, Milligan BG. 1994. Analysis of population genetic structure with RAPD markers. Molecular Ecology, 3: 91-99. [ Links ]
12. Mantel NA. 1967. The detection of disease clustering and a generalized regression approach. Cancer Research, 27: 209-220. [ Links ]
13. Miller MP. 1997. Tools for population genetic analyses (TFPGA). Version 1.3. Computer software distributed by author. [ Links ]
14. Nei M. 1973. Analysis of gene diversity in subdivided populations. Proceeding of the National Academy of Science, 70: 3321-3323. [ Links ]
15. Nei M. 1978. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics, 89: 538-590. [ Links ]
16. Peakall R, Smouse PE. 2006. GENALEX 6: genetic analysis in Excel. Population genetic, software for teaching and research. Molecular Ecology Notes, 6: 288-295. [ Links ]
17. Rohlf FJ. 2002. NTSYS pc: Numerical taxonomy system, Version 2.1. Setauket (New York, U. S. A.): Exeter Publishing. [ Links ]
18. Sánchez MD. 2000. Mulberry, an exceptional forage available almost worldwide. [Internet], Fecha de acceso: 2012 enero 23. Disponible en: http://www.fao.org/ag/AGA/AGAP/FRG/Mulberry/Papers/HTML/Mulbwar2.htm. [ Links ]
19. Shabir AW, Bhat MA, Malik GN, Kamili AS, Mir MR, Bhat SA, Wani N, Razvi SM, Akhtar S, Bhat KA. 2010. Molecular markers and their role in mulberry improvement. International Journal of Current Research, 4: 20-24. [ Links ]
20. Sharma A, Sharma R, Machii H. 2000. Assesment of genetic diversity in a Morus germplasm collection using fluorescence-based AFLP markers. Theoretical and Applied Genetics, 101: 1049-1055. [ Links ]
21. Sneath PHA, Sokal RR. 1973. Numerical taxonomy, the principles and practice of numerical classification. San Francisco (U. S. A.): W. H. Freeman & Co. p. 573. [ Links ]
22. Sokal RR, Rohlf FJ. 2012. Biometry: the principles and practice of statistics in biological research. 4th ed. New York (U. S. A.): W. H. Freeman and Co. p. 937. [ Links ]
23. Thome J, Gonzáles DO, Beebe S, Duque MC. 1996. AFLP analysis of gene pool of a wild bean core collection. Crop Science, 36: 1375-1384. [ Links ]
24. Vijayan K, Chatterjee SN. 2003. ISSR profiling of Indian cultivars of mulberry (Morus spp.) and its relevance tobreeding programs. Euphytica, 131: 53-63. [ Links ]
25. Vijayan K, Nair CV, Chatterjee SN. 2009. Diversification of mulberry (Morus indica var. S36), a vegetatively propagated tree species. Caspian Journal of Enviromental Science, 7 (1): 23-30. [ Links ]
26. Vijayan K, Saratchandra B, Teixeira da Silva JA. 2011. Germplasm conservation in mulberry (Morus spp.). Scientia Horticulturae, 128: 371-379. [ Links ]
27. Vos P, Hogers R, Bleeker M, van der Lee T, Hornes M, Frijter A, Pot J, Kuiper M, Zabeau M. 1995. A new technique for DNA fingerprinting. Nucleic Acid Research, 23: 4407-4414. [ Links ]
28. Wang ZW, Yu MD. 2001. AFLP analysis of genetic background of polyploid breeding materials of mulberry. Acta Sericologica Sinica, 27 (3): 170-176. [ Links ]
29. Wright S. 1931. Evolution in Medelian populations. Genetics, 28: 114-138. [ Links ]
30. Xiang ZH, Zhang Z, Yu MD. 1995. Preliminary report on the application of RAPD in systematics of Morus L. Acta Sericologica Sinica, 21 (4): 204-208. [ Links ]
31. Yamanouchi H, Koyama A, Takyu T, Muramatsu N. 2010. Nuclear DNA amounts in diploid mulberry species (Morus spp.). Journal of Insect Biotechnology and Sericology, 79 (1): 1-8. [ Links ]
32. Yang G W, Feng L C, Jing C J. 2003. Analysis of genetic structure variance among mulberry (Morus spp L.) Populations. Acta Sericologica Sinica. 29 (4): 323-329. [ Links ]
33. Yeh FC, Yang RC, Boyle TBJ, Ye ZH, Mao JX. 1997. POPGENE, the user-friendly shareware for population genetic analysis. Canadá: Universidad de Alberta. [ Links ]
34. Zhao WG, Pan YL, Huang MR. 2000. RAPD analysis for the germplasm resources of genus mulberry. Acta Sericologica Sinica, 4: 1-8. [ Links ]
35. Zhao WG, Pan YL. 2004. Genetic diversity of genus Morus revealed by RAPD markers in China. International Journal of Agriculture y Biology, 6: 950-954. [ Links ]
36. Zhao WG, Miao XX, Pan YL, Huang YP. 2005. Isolation and characterization of microsatellite loci from the mulberry, Morus L. Plant Science, 168 (2): 519-525. [ Links ]
37. Zhao WG, Miao XX, Zang B, Zhang L, Pan YL, Huang YP. 2006. Construction of fingerprinting and genetic diversity of mulberry cultivars in China by ISSR markers. Acta Genetica Sinica, 33 (9): 851-860. [ Links ]
38. Zietkiewicz E, Rafalski A, Labuda D. 1994. Genome fingerprinting by simple sequence repeat (SSR)- anchored polymerase chain reaction amplification. Genomics, 20: 176-183. [ Links ]

