











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCTION
Circular RNAs (circRNAs) are part of a new class of RNAs described as non-coding. Previously, they were known as reject molecules generated by an aberrant RNA splice or specific to some pathogens (Cocquerelle et al., 1993) such as the hepatitis virus δ (Kos et al., 1986) and some plant viroids (Sanger et al., 1976). However, a big number of circRNAs are endogenously expressed in mammalian cells, many of which were found to be abundant and stable, with multiple functions (Memczak et al., 2013). CircRNAs can arise from exons only (exonic circRNA), introns only (intronic circRNA), or a combination of both, with independent generation modes (Zhang et al., 2013).
Currently, there is growing interest in circRNAs, not just because of their unique biogenesis mechanisms, but also on their roles in cellular pathways, disease pathologies, and usefulness as molecular markers (Figure 1).
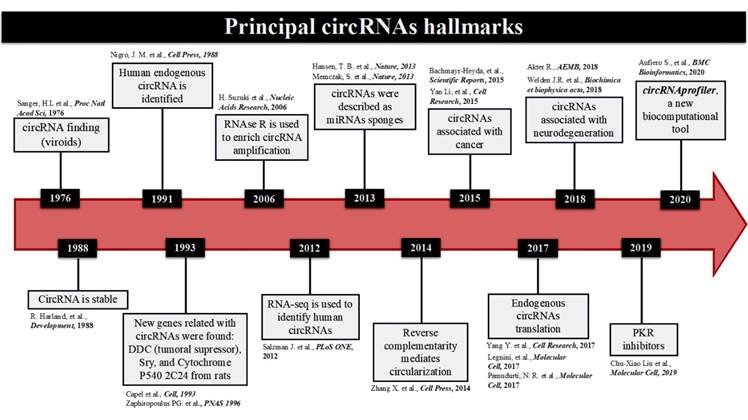
Figure 1 Timeline of the principal circRNAs Hallmarks. A brief description of circRNAs discovery, going through their structural stability, first enrichment techniques, factors that favored their biogenesis, functions as miRNAs sponges, pathologies association, their first endogenous translation, and the complete bioinformatic tool to predict not only location, but also interactions with other genes pursuing functionality in the cell.
CircRNA biogenesis and detection
The biogenesis of circRNAs suggests they are formed from backsplicing, which occurs after alternative splicing and consists of intronic bonds formation altering exon order, leading the subsequent exon circularization by joining the donor site with the acceptor site (Jeck & Sharpless, 2014), generating an RNA molecule with a circular structure due to a covalent bond between its 5' and 3' ends. This structure is stabilized by a phosphodiester bond between the donor and acceptor sites which makes it more stable than linear RNAs.
Recent research on circRNA biogenesis has shown that backsplicing is catalyzed by the canonical splicing machinery (Ashwal-Fluss et al., 2014). While most eukaryotic circRNAs are generated by the spliceosome, yeast and plants circRNAs have not-so-similar mechanism. In general, circRNA biogenesis in plants is mediated by extensive introns without great complementarity between each other (Sun et al., 2016; Ye et al., 2015). Whereas in Archaea, its circRNA formation is not related with the spliceosome (Salgia et al., 2003). Long flanking introns in human genes have been observed surrounding specific circularized exons of Deleted in Colorectal Carcinoma (DCC) genes and transcription factor EST-1 during splicing (Cocquerelle et al., 1992). This study also showed mostly circRNAs located in the cytoplasm and their stability 48 hours post cell treatment with actinomycin D, confirming their great structural resistance compared with linear RNAs.
In 1995, it was hypothesized that inverted repeats inside flanking introns were necessary for circular RNA formation (Dubin et al., 1995); describing also that within these long flanking intron sequences, ALU-SINEs in humans and like ALU in mice, can be found (Jeck et al., 2013) (Figure 2).
It is known that these ALU sequences, besides introducing new splicing sites, to generate new mRNA isoforms transcription, are also complementary to each other, forming structures known as Inverted Repeated Alu elements (IRAlus). This IRAlus formation turns these ALU sequences into potential RNA regulators and participants in circRNAs biogenesis (Chen & Carmichael, 2008; Kreahling & Graveley, 2004).
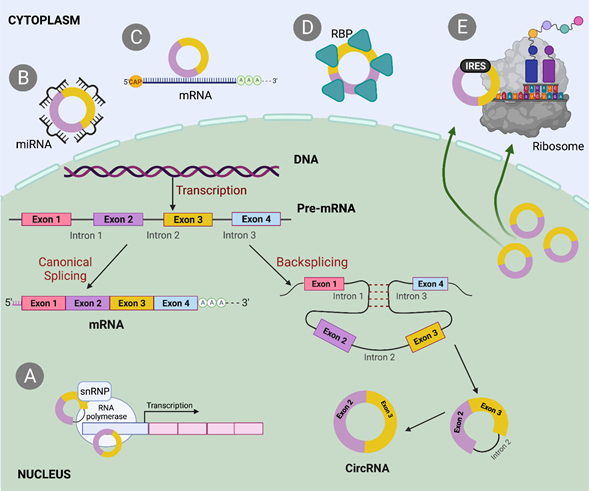
Figure 2 Biogenesis and mechanisms that involved circRNAs. CircRNA biogenesis mediated by backsplicing and driven by ALU sequences localized inside intronic regions. A. When circRNA localized in the nucleus can interact with the transcription process of its own gene. B. CircRNA interacts with microRNAs up or down-regulating different pathways in which miRNAs are involved. C. As part of competition mechanism, circular RNAs can interact directly with its own messenger causing mostly the decay of this linear counterpart. D. CircRNAs can act as RBP sponges and gain functionality. E. Some circular RNAs have IRES, which facilitate the interaction with ribosome and translation.
The identification and quantification of these circular isoforms require a specific bioinformatic design which starts with access to circRNAs databases. Since circRNAs were found, several bioinformatic tools have been development to predict either biogenesis or some specific functions. Currently, these tools can even design specific primers for their detection and amplification through PCR technique (Xu, 2017), highlighting the fact that the primers orientation should always be divergent. Since the RNA-seq was complemented with RNAse R pre-treatment and the depletion of poly-A tail, this helped to enrich circRNAs due to their increased RNAse resistance. Another technique called CircRNA-seq has been used to provide more reliability to the approaches, adding other tools to detect alternative splicing events, circRNA structures, and statistical testing of candidates to stablish an interaction with other target molecules (Jakobi & Dieterich, 2019).
On the other hand, backsplicing seems to be regulated by cis-acting and trans-acting splicing factors through binding to specific motifs of flanking introns to generate circRNAs from pre-mRNAs. Although few studies have conclusively demonstrated a direct interaction between a splicing factor and pre-mRNA. As an example of this, ADAR proteins bind to a double stranded RNA to deaminate adenosine and mediate the release of RNA (Nishikura, 2010). This model suggests that inverted repeats are destabilized in the presence of ADAR, inhibiting the circRNA splicing and when ADAR silencing is performed, greater circRNA expression is generated (Ivanov et al., 2015). However, it was not demonstrated that circRNA expression is only regulated by ADAR activity.
CircRNA competition with mRNA splicing
Most circRNAs are generated from protein-coding genes, containing in some cases a big number of exons. This suggests that circRNAs have been transcribed by RNA polymerase II (RNA pol II) and that their biogenesis may be spliceosome mediated. RNA pol II influences pre-mRNA processing through changes in the elongation rate, acting as a modulator of alternative splicing (Jeck et al., 2013; Kornblihtt et al., 2013).
It is also known that circRNAs are typically generated at expense of canonical isoforms of mRNAs and compared to these, the circular molecules are even less abundant. However, their biogenesis may regulate the expression of their pre-mRNA precursor, where circRNAs and mRNAs generation are competing. This competence is tissue-specific and, in some cases, conserved between flies and humans (Salzman et al., 2012).
Regarding this tissue-specific competence, a group of researchers showed that either in Drosophila and the vertebrate counterpart, the Muscleblind splicing factor (MBL) and Muscleblind-like protein 1 (MBNL1), respectively, have the second exon circularized, and this circRNA contains a start codon. This exon is flanked by long intronic sequences where both, circRNA and flanking introns, contain highly conserved Muscleblind binding motifs, which specifically bind this MBL RNA to its circRNA counterpart. This research proves that circRNAs transcription rate is determined mainly by flanking introns, supporting previous data suggesting the presence of ALU sequences within long introns would lead to a circularization of exons or introns during the splicing process, determining a factor for biogenesis of circular RNAs (Ashwal-Fluss et al., 2014; Jeck et al., 2013).
Concerning tissue specificity and circRNAs production based on stages of development, independent studies showed that many circRNAs are abundant in brain and their levels increase during differentiation and neuronal development (Rybak-Wolf et al., 2015; Venø et al., 2015; Westholm et al., 2014; You et al., 2015). In contrast, it is suggested the production of circRNAs is modulated by neuronal activity and enriched in synaptosomes and dendrites (You et al., 2015). This cumulative presence in neural tissue was most evident during aging (Cortés-López et al., 2018; Gruner et al., 2016; Westholm et al., 2014), which would be correlated with low rate of cell regeneration in neural tissue.
Role of RNA-binding proteins (RBPs) in circRNAs synthesis
After finding that MBL protein acts as direct intermediary in circMBL synthesis through binding to its messenger, some RBPs were identified regulating the expression of different circRNAs in different systems and organisms.
Such RBPs include Quaking Homolog, KH Domain RNA Binding (QKI), FUused in Sarcoma/Translocated in LipoSarcoma (FUS), Nuclear Factors NF90/NF10, DExH-Box Helicase 9 (DHX9), splicing regulatory Protein1 (ESRP1), and proteins rich in Serine/arginine (SR) (Conn et al., 2015; Errichelli et al., 2017; Yu et al., 2017). QKI can promote circRNA biogenesis during the transformation of epithelium to mesenchyme in human cell culture, since this circularization of exons would be mediated by the presence of binding sites within the flanking intronic sequences (Conn et al., 2015).
The RBP FUS was found mediating circularization in murine embryo cells by specific junction site between exon-intron junctions (Errichelli et al., 2017). The same year, the splicing factor ESRP1 was reported as a circularization mediator for circBIRC6 (a baculoviral derivative which contains six genes), this was explained by the presence of a specific binding site in one of the introns flanking the circularized exon (Yu et al., 2017).
That information relates RBPs to specific binding sites, predominantly in intronic flanking regions or junctions between exons and introns, allowing complementarity between intronic sequences, bringing on the exon circularization. This event results in a high rate of circRNA transcription, which means that circRNAs are not only reject molecules. Instead, this high translation rate has allowed to explore circRNAs and evaluate their cellular regulatory functions.
CircRNA functions
There have been numerous speculations on the functions of circRNAs. In 2013, it was thought that circRNAs could act as microRNA (miRNAs) sponges. This concept emerged from the observation when two circRNAs (CDR1as known as ciRS-7 and circSry) were detected bounded to miRNAs and modulating their specific activities (Hansen et al., 2013; Memczak et al., 2013).
Experiments with circRNAs have shown that they can act as microRNAs sponges, and thus regulate some signaling pathways in cancer. The function of circRNAs as microRNA sponges was further supported through RNA analysis, comparing linear RNAs expression against the circular species in normal colon mucosa, tumoral tissues, and colorectal cancer cell lines, confirming circRNA expression ratio was negatively correlated with the proliferation rate in tumoral tissue and cancer cells (Bachmayr-Heyda et al., 2015). These findings strongly support the notion that circRNAs are highly dynamic molecules with biogenesis being both intricately regulated and cell type dependent. The role of circRNAs as negative regulators of the microRNAs provides a functionality that changes its status from "disposal molecule."
Driven by the need for circRNA prediction methods, a new computational tool named “circRNAprofiler” was designed using other platforms mentioned previously. This program includes different techniques interacting one to each other and distributed in 15 different modules as: a project folder containing a genome-annotation file, with experimental design and prediction results, followed by merging this information with annotations from other alternative tools with the objective to create a unique data set for downstream analyses, eliminating false positives through the R Bioconductor packages as DESeq2 and EdgeR, which are used to model changes in circRNAs expression. The next modules are focused on Back Spliced Junctions (BSJ) to enrich the analysis in microRNA/RBP binding sites, extracting the internal circRNA sequences, but also analyzing the flaking intronic sequences, looking for SNPs which can affect circRNAs biogenesis. This framework provided by circRNAprofiler might be the most complete computational tool for the study of circRNA to date. The platform provides elements to study the expression, function, and biogenesis of circRNAs across any physiological or pathological entity (Aufiero et al., 2020).
MicroRNA sponge
Although circRNAs functions remain largely unknown, research has shown they can act as miRNA sponges, regulating different signaling pathways. For example, exogenous circRNA CDR1as/CiRS-7 from the CDR1 gene encodes a protein related to cerebellar degeneration and acts as a potent sponge/inhibitor of micro-RNA-7 (miR-7), affecting the expression of this microRNA in the Central Nervous System (CNS) (Memczak et al., 2013; Salgia et al., 2003). The role of miR-7 as a key regulator of several oncogenic cascades and its implication in Parkinson's disease makes CDR1as/CiRS-7 a crucial factor for neurons functioning, as well as a responsible candidate in neurological disorders and brain tumor development (Geng et al., 2016; Hansen et al., 2013).
Another example is the circRNA circ-HIPK3 generated from the second exon of HIPK3 mRNA encoding a protein kinase mostly related to cell senescence. This circRNA can work as a sponge of nine miRNAs, including miR-124, and inhibit their activity. It was also shown silencing circHIPK3 reduces cell proliferation by affecting the presence of miR-124 through its sponge function, and consequently regulating the levels of its target mRNAs (Zheng et al., 2016).
CircRNAs involved in signaling pathways
Several findings indicate that circRNAs can modulate cell fate since they are involved in different signaling pathways acting as both agonists and antagonists of proteins. Using bioinformatic tools such as RBP database, it has been demonstrated that circRNAs can bind or sequester RBPs, however these circRNAs have a lower RBP binding density when compared to mRNAs (You et al., 2015). A clear example is circ-Foxo3, derived from the transcription factor Foxo3. Circ-Foxo3 was highly expressed in non-cancerous cells, and its presence was associated with cell cycle progression. Circ-Foxo3 silencing promotes cell proliferation, and its ectopic expression represses cell cycle mediated by its binding to CDK2, a cyclin 2-dependent kinase protein, and p21, a cyclin 1A-dependent kinase inhibitory protein. This double interaction results in the formation of a ternary RNA-protein complex (circ-Foxo3-p21-CDK2) that represses cell cycle proliferation and progression in tumor cells (Du et al., 2016).
Upon further research, it was determined circ-Foxo3 promoted cardiac senescence by modulating factors associated with stress responses and senescence. Circ-Foxo3 is highly expressed in heart samples from elderly patients and adult mice where doxorubicin (Dox)-induced cardiomyopathy is aggravated by its ectopic expression, while relieved by silencing endogenous circ-Foxo3 (Du et al., 2017). These studies demonstrated that circ-Foxo3 is mainly distributed in the cytoplasm, interacting with ID-1, an anti-senescent protein, and the transcription factor E2F1 and anti-stress proteins FAK and HIF1α. The authors conclude that the interaction of ID-1, E2F1, FAK, and HIF1α with circ-Foxo3, retains these proteins in the cytoplasm, inhibiting their anti-senescent and anti-stress functions, resulting in increased cell senescence.
Another example for this circRNAs interaction with signaling pathways was shown in the expression of circ-Amotl1, derived from the Amotl1 gene which regulates endothelial cell migration and capillary formation. After microarray analysis of neonatal and postnatal human cardiac tissue samples, circ-Amotl1 was highly expressed in neonate cardiac tissue. Circ-Amotl1 was overexpressed in endothelial cell line YPEN and human MCF-7 cells, showing that circ-Amotl1 can enhance the survival of cardiomyocytes by binding to PDK1 and AKT1 protein kinases, phosphorylating AKT1, and facilitating its translocation to the nucleus to promote cell survival. In vivo studies proved circ-Amotl1 overexpression is protective against Dox-induced cardiomyopathy and can counteract the damage generated by Dox (Zeng et al., 2017). This work supports the direct interaction between circular RNAs, and protein kinases involved in cell survival or cell cycle arrest, directly tissue dependent.
CircRNAs degradation
The stability of circRNAs was confirmed after RNAse R treatment, a 3'-exonuclease capable of degrading linear RNAs. These circular molecules resisted degradation due to their closed-loop structure by 5' and 3' covalent bonds that confer stability against enzymatic degradation. Therefore, RNAse R is useful to enrich their presence, and the differentiation of linear mRNAs with scrambled exons (Suzuki et al., 2006).
In some studies, circRNAs have been observed accumulated in brain tissue, inferring that their hoarding rate varies according to the tissue type. In addition, circRNAs have been seen to pile up predominantly in tissues with low cell division, combining information that refers to an age-dependent accumulation in the brain (Ashwal-Fluss et al., 2014).
Furthermore, in 2017 it was shown that circRNAs are less expressed in tissues with a high rate of cell proliferation, such as epithelial-ovarian and hepatic (Bachmayr-Heyda et al., 2015). The reduced expression in this type of tissue suggests that cell division decreases the transcription rate of this circular molecules and consequently its expression levels.
In 2018, the GW182 protein was reported as a scaffold recruited by argonauts to form part of miRNAs repression complex, inducing downstream translational repression, and facilitating the formation of P-bodies (Niaz & Hussain, 2018). Next, GW182 was shown to play a direct role not only in miRNA degradation, but also in the modulation of circRNA expression. They found that GW182 depletion, increased the transcriptional levels of circ-dapi and circ-laccase2 in Drosophila. The circular RNA circ-laccase2 was 10 times more abundant than its linear isoform, demonstrating that GW182 had a strong potential to affect the stability of mature circRNAs, but not nascent circRNAs, therefore, it would not be affecting their biogenesis. It was also found that proteins TNRC6A, TNRC6B, and TNRC6C have a 39% homology to GW182. These three homologs were silenced using siRNAs in HEK293 cells, resulting in 12 circRNA with different lengths and number of exons. The circRNAs were increased in the absence of these homologous proteins, which would indicate that GW182 does indeed have a conserved and essential role in the degradation of some circRNAs (Jia et al., 2019).
It is known GW182 protein has a middle domain that works as a scaffold and makes possible the recruitment of some RNA disintegration factors (Niaz & Hussain, 2018); therefore, through this middle domain, GW182 would be regulating factors involved in circRNAs degradation.
Transport of circRNAs from the nucleus to the cytoplasm
Although it is known most mature circRNAs are in the cytoplasm, it is essential to mention some intronic and intron-exonic circRNAs are retained in the nucleus as they can regulate the transcription of the parental genes (Li et al., 2015; Patop et al., 2019).
The fact circRNAs are primarily found in the cytoplasm is not surprising since different RNAs are exported from the nucleus to the cytoplasm by a specific route to fulfill a particular role. In the canonical path of mRNAs, the transcript is exported from the nucleus to be translated into proteins at the ribosome, that is why is important to understand how this transport mechanism of circRNAs works.
The transportation of circular RNAs from the nucleus is dependent on molecule length and this system is preserved in Drosophila. With the purpose to identify the factors involved in circRNA, 26 cytoplasmic export proteins involved in RNA nuclear to cytosolic transport were inhibited. It was reported the Helicase Hel25E deletion results in nuclear accumulation of circRNAs with lengths greater than 800 nucleotides, but not directly in the short length circRNAs. In humans, the Hel25E homologs have similar regulatory localization activity, since when the UAP56 and URH49 deletion was evaluated, the nuclear localization of longer and shorter circRNAs was enriched, respectively (Huang et al., 2018; Li et al., 2019).
These cumulative discoveries suggest mature circRNAs are being exported from the nucleus to the cytoplasm is a length dependent manner.
Pathologies in which different circRNAs have been implicated
Proving their viability as molecular indicators; circRNAs were reported abundantly in spit and blood samples, and currently are used as clinical biomarkers for disease prognosis and progression (Jeck et al., 2013; Lee et al., 2019).
It was shown the circular Heart-related (HRCR) works as anti-hypertrophic molecule acting as a protector against heart failure, this was evidenced with HRCR working as miRNA-223 sponge, an endogenous microRNA that induces cardiac hypertrophy (Taïbi et al., 2014).
Previously, it was described that circRNAs have been primarily found in brain tissue showing different expression profiles at the hippocampus during its maturation, suggesting their implication in development of some pathologies such as Alzheimer's disease (AD), Parkinson's disease (PD) and cerebral ischemia (Ashwal-Fluss et al., 2014; Rybak-Wolf et al., 2015).
The miR-7 is a stable and abundant molecule in the CNS acting as an essential post-transcriptional regulator of human gene expression in the brain. Endogenous circRNA (ciRS-7) is also expressed in this tissue and has different recognition motifs for miR-7, forming a miR-7/ciRS-7 complex that turns off the functions of the microRNA. Currently, it is known that in the hippocampus region of spontaneous AD patients, the miR-7/ciRS-7 complex is deregulated, which significantly reduces miR-7 levels; moreover, when the miRNA is overexpressed, ciRS-7 levels decrease.
This variation in miRNA levels has repercussions on the deregulation of targets, such as the ubiquitin ligase A protein (UBE2A), which can interact with miR-7 and acts as a phagocytic amyloid protein in brains with AD (Akhter, 2018; Lukiw, 2013).
Even when mutations in Tau protein involved in microtubules association (MAPT) are described as responsible for the development of different neurodegenerative diseases, referred to as “taupathies”, which includes AD and frontotemporal lobar degeneration/frontotemporal dementia (FTLD/FTD), their mechanisms remain unclear. Through PCR screen using human brain RNA and using divergent primers plus RNAse R to avoid linear RNAs amplification, two Tau circRNAs were reported through exon 12 backsplicing to either exon 10 or 7. Even when the 12 to 10 circRNA did not contain a start codon, it was the most abundant. To investigate this, a minigene technique was performed and transfected into HEK293 cells to detect this 12 to 10 circRNA. The minigene contained repetitive elements surrounding exon 12 and an intronic repetitive element upstream of exon 9, due to the presence of ALU elements that stimulate circRNAs formation. Despite the weak expression of this Tau circRNA in transfected cells, it was reported that the exon 12 to 10 circular is regulated by cdc2-like kinase (CLK2) through trans-acting factor expression. Since it contains the exon 10, which is alternatively spliced in linear MAPT, the circRNA is sensitive to mutations present in this exon, these mutations promote exon 10 inclusion, influencing in tau 12 to 10 circRNA formation. Despite the absence of a start codon in Tau 12 to 10 circRNA, its sequence is divisible by three, theoretically suggesting an alternative mechanism of edition made by ADAR1, capable to change an AUA to AUG triplet (start codon). This editing effect does not exclude a possible translation that can have an effect in AD, either its brain accumulation as a circRNA or as a new peptide with functional characteristics (Welden et al., 2018).
Parkinson's disease (PD) is known for affecting dopamine-producing neurons in a specific area of the brain. Additionally, miR-7 can inhibit the expression of α-synuclein, which is highly expressed in PD patients, and its detection is used as a marker of the disease. Furthermore, this negative regulation of α-synuclein by miR-7 protects cells against oxidative stress and neuronal death caused by MPP+, a neurotoxin that interferes with oxidative phosphorylation in mitochondria (Choi et al., 2014; Junn et al., 2009; Shao & Chen, 2016).
In 2018 it was demonstrated that circSNCA derived from the SNCA gene (α-synuclein) is associated with reduced expression of pro-apoptotic proteins (CASP3, BAX, PTEN, and P53) and its negative regulation with pramipexole (PPX) would decrease apoptosis and induce autophagy in PD, acting as a miR-7 sponge to regulate the levels of α-synuclein (Sang et al., 2018).
Since there are many circRNAs recently described in other pathologies such as cancer, some regulatory functions have been studied in specific signaling pathways.
In gastric cancer (GC), 201 circRNAs were found downregulated, while 107 circRNAs were over-regulated. Within these dysregulated circRNAs, hsa_circ_0014717 stood out being found in 77.2% (74/96) of tissues with CG and associated with tumor stage, distant metastasis, and expressions of tumor biomarkers, including CA19-9 and CEA (Ning et al., 2018).
Liver cancer was another pathology found to be modulated by circRNA activity. Young hepatoblastoma tissues were analyzed by circular RNA microarray, whereby RNA isolation was complemented with RNAse R to enrich circular transcripts, adding verification through qRT-PCR. The outcome showed some up regulated and significantly expressed circRNAs which were selected. Using bioinformatic tools for the annotation and functional analysis as Gene Ontology and KEGG pathway analysis, potential circRNAs capable to bind miRNAs were predicted. This is the case of circ_0015756, that was significantly up-regulated in hepatoblastoma specimens and metastatic hepatoblastoma cell lines, is also capable to act as miR-1250-3p sponge, regulating its function in the development of hepatoblastoma. Therefore, when circ_0015756 was silenced in vitro, proliferation, cell invasion, and viability of hepatoblastoma decreased (Liu et al., 2018).
It is important to highlight the capacity of circRNAs to function as modulators for different signaling pathways, through the interaction with miRNAs or RBPs. In addition, being used as biomarkers to determine the presence of specific diseases, circular RNAs allow the activation or deactivation of proliferative pathways within different pathologies, becoming a new molecular tool to be used in cancer screening. Circular RNAs work as regulators in neurodegenerative diseases, correlating their expression/accumulation not only with aging, also showing participation with alternative splicing and ALU sequences dependence.
Circular RNAs translation
General translation of mRNA begins when the translational machinery recognizes the end 5' CAP and scans the UTR region until the start codon is recognized. Translation can also occur independently of the 5'CAP and requires an internal ribosome entry site (IRES). It was unknown whether the messenger translational machinery acted on molecules with such a particular structure as circRNAs. These are generally not associated with polyribosomes, suggesting that circRNAs do not translate (Jeck et al., 2013).
Several circRNAs have IRESs, prompting they could be translated into small proteins or micro peptides, also showing an in vitro synthesized circRNA containing an IRES, binds to the 40S ribosomal subunit and initiates translation (Dudekula et al., 2016).
A recent publication supports the translation of circRNAs by a specific association with active ribosomes by generating proteins from circRNAs. Using the Ribosome Foot Printing (RFP) technique in Drosophila to determine which types of messengers are actively translating, as evidence of a stop codon was found in circMbl. Further investigation demonstrated the presence of sequences that promote cap-independent translation of different circRNAs. From this analysis, a peptide coinciding with circMbl3 (an isoform of circMbl) was also obtained, for which no known linear variant was found. Also, putative proteins were found encoded by circMbl using western blot technique and determining also physiological factors that can regulate the levels of these proteins (Pamudurti et al., 2017).
CONCLUSIONS
The studies described in this article suggest circRNAs can act in different ways as positive or negative regulators of the cellular environment having a big impact on cell function, discrediting the idea of circRNAs being only accumulative and non-functional molecules.
Although no clinical trials of circRNAs as therapeutic target in human diseases have been reported yet, it is likely circRNAs are recruiting other RNAs and generating different effects. Through circRNA binding to microRNAs, they can regulate transcriptional and translational processes. The stability and possible sequence modification of circular RNAs mediated by ADAR1 increase the potential to generate functional peptides. These circRNA peptides could share the same protein activity with the canonical isoform or even as a protein sponge for the encoding own gene.
Using circRNAs as biomarkers would allow the rapid analysis of patients with different pathologies without subjecting them to a largely invasive sampling procedure. In addition, the amount of bioinformatic tools currently available assist the identification circRNAs involved in multiple mechanisms, simplifying their study, and providing knowledge about their specific functions. These functions can be tissue-specific and dependent on the stage of cell development. We believe studying these molecules as signaling agents in high impact pathologies will allow the development of novel diagnostic and therapeutic regimes.

