










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista de la Academia Colombiana de Ciencias Exactas, Físicas y Naturales
versão impressa ISSN 0370-3908
Rev. acad. colomb. cienc. exact. fis. nat. vol.35 no.136 Bogotá jul./set. 2011
BIOQUÍMICA
OBTENCIÓN DE UN SUB-PROTEOMA DE CITOPLASMA DE UNA LÍNEA CELULAR DE TROFOBLASTO MEDIANTE FRACCIONAMIENTO CON DETERGENTES
1 Estudiante de doctorado.
2 Profesor Titular. Grupo de Investigación en Hormonas, Departamento de Química, Universidad Nacional de Colombia. Cra. 30 45-03, Bogotá D.C., Colombia. Correo electrónico: mysanchezd@unal.edu.co.
* Autor para correspondencia.
Resumen
El método de fraccionamiento con detergentes descrito por Rambsy y Makowski en 1999, fue modificado utilizando un buffer con digitonina (0,015%) para permeabilizar células HTR8/SVneo de trofoblasto humano, extrayendo las proteínas de citoplasma en la fracción soluble y solubilizando las de membrana a partir del precipitado con un buffer con Tritón X-100 (0,5%). Se obtuvo un extracto reproducible de proteínas citoplasmáticas libre de proteínas de membrana y se adecuó para electroforesis bidimensional (2-DE) por precipitación con clorofomo/metanol. En conclusión, el método aquí descrito resulta adecuado para la obtención de sub-proteomas de citoplasma a partir de líneas celulares, con una reproducibilidad y recuperación óptimas para el análisis de perfiles proteómicos por electroforesis bidimensional.
Palabras clave: Proteómica, fraccionamiento celular, Tritón X-100, digitonina, trofoblasto, HTR8/SVneo.
Abstract
The differential detergent fractionation method described by Rambsy and Makowski in 1999, was modified using a digitonin (0.015%) buffer to permeate HTR8/SVneo human trophoblast cells and obtain the cytoplasm proteins in the soluble fraction, while membrane proteins were dissolved from the pellet with Triton X-100 (0.5%) buffer. We obtained a reproducible membrane-free cytoplasm protein extract suitable for 2-DE, by chloroform/methanol precipitation. In conclusion, the method here described is appropriate for the separation of cytoplasm sub-proteomes from cell cultures, with an optimal reproducibility and recovery to perform proteomic profile analysis by bidimensional electrophoresis.
Key words: Proteomic, cell fractionation, Triton X-100, digitonine, trophoblast, HTR8/SVneo.
Introducción
Durante el primer trimestre de embarazo se desarrolla el trofoblasto como parte importante de la placenta. El trofoblasto, especialmente el citotrofoblasto extravelloso (EVT), comparte muchas características con células cancerosas, como el alto nivel de proliferación, migración e invasión; sin embargo, la función del trofoblasto se encuentra altamente controlada a diferencia de las células malignas (Ferretti, Bruni et al. 2007). por lo anterior, es de interés estudiar el perfil de proteínas expresadas por el trofoblasto, con miras a identificar la desregulación que tiene lugar en el desarrollo maligno. Solo un enfoque proteómico permite captar la globalidad del sistema y la detección de proteínas hasta ahora no reportadas en este modelo.
La proteómica es una disciplina que ha emergido dentro de las técnicas bioquímicas para ayudar a revelar los mecanismos bioquímicos y fisiológicos celulares y en particular de enfermedades complejas, donde se permite la comparación cualitativa y cuantitativa de todas las proteínas expresadas por una célula bajo determinadas condiciones. La electroforesis bidimensional es una de las técnicas de proteómica más usada actualmente para la separación de mezclas complejas de proteínas, la cual en asocio con técnicas de análisis de imágenes e identificación de proteínas por espectrometría de masas, permite analizar un perfil proteico particular (Reiner Westermeier, 2008). No obstante, la complejidad de las muestras y el rango dinámico tan amplio de los extractos proteicos representa un desafío analítico a la hora de separar y analizar proteomas.
Las proteínas de membrana desarrollan un papel muy importante en señalización celular, adaptación a ambientes, e interacciones célula-matriz y célula-célula, y además, están probablemente involucradas en la adquisición y mantenimiento de propiedades metastásicas e invasivas de células tumorales. Las proteínas de membrana representan más del 50% de los principales blancos terapéuticos actuales. Son pocos los estudios desarrollados sobre proteínas de membrana asociadas a cáncer, dentro de ellos se pueden destacar los siguientes: cáncer de seno (Liang, 2006; Dowling, 2007), carcinoma nasofaringeal (Zhang, 2008), tejidos de tiroides malignos y benignos, células no cancerosas como las células madre mesenquimales humanas (Foster, 2005), neutrófilos (Jethwaney, 2007) y queratinocitos (Gonnet, 2003). Dichos estudios han reportado la expresión diferencial de proteínas o la identificación de todas las proteínas de membrana extraídas. Sin embargo, el estudio de proteínas de membrana es un desafío, presentando dos problemas principales: su baja abundancia relativa y su pobre solubilidad en medios acuosos (Vertommen, Panis et al. 2011).
Para analizar mejor un proteoma es necesario realizar un fraccionamiento previo, con el que se logre reducir la complejidad de la muestra, mejorando la separación y evitando la superposición de proteínas abundantes, y en consecuencia incrementando considerablemente el número de identificaciones. Existen diferentes técnicas para realizar este fraccionamiento, la mayoría orientadas a extractos bacterianos o con pasos que involucran ultracentrifugación (Payrastre, Plantavid et al. 1988; Selinsky 2003; Roobol- Bóza, Dolby et al. 2004; Everberg, Leiding et al. 2006), pero son pocas las que se refieren a extractos de líneas celulares humanas. Dentro de las técnicas que permiten trabajar células eucariotas, está la desarrollada por Rambsy y Makowski en 1999 (Walker, 2005). Las ventajas de este método es que es simple y altamente reproducible, no requiere pasos de ultra centrifugación, preserva la integridad de los filamentos de la red del citoesqueleto y es especialmente útil para cantidades limitadas de biomaterial; además, se obtiene cada fracción de forma soluble y no precipitada, cuya solubilización es causa de problemas, especialmente para proteínas de membrana (McCarthy, Burgess et al. 2005). Este método fue implementando en este trabajo, y con el objetivo de mejorar la recuperación de proteínas se modificaron los tiempos de incubación, y se implementaron una serie de lavados tendientes a la limpieza de cada fracción y a aumentar la recuperación de proteína.
Otro punto importante en la obtención de proteomas, es la limpieza de la muestra previa al análisis por isoelectroenfoque. Es necesario eliminar el exceso de sales y lípidos, así como concentrar la muestra y disponerla en el buffer de isoelectroenfoque, para que esta pueda ser correctamente separada en función de su punto Isoeléctrico (pI). Existen diversas técnicas de purificación por precipitación; aunque en general se tiene una baja recuperación, es necesario verificar que no haya falta de reproducibilidad ni una pérdida selectiva o heterogénea de proteína durante estas pérdidas.
Debido a que la línea de trofoblasto, HTR8/SVneo, es un modelo interesante para comprender la patología del cáncer, la obtención de sub-proteomas de membrana y de citoplasma y su análisis por electroforesis bidimensional es de gran interés, siendo este el objetivo del presente trabajo.
Materiales y métodos
Cultivo celular
La línea celular de citotrofoblasto extravelloso HTR8/ SVneo fue donada por la Dra. Ángela Cadavid de la Universidad de Antioquia. Esta línea celular fue desarrollada de un cultivo de explante de placenta humana de primer trimestre e inmortalizada por transfección con un ADNc que codifica el antígeno T 40 grande del virus de simio (Graham, Hawley et al. 1993). Estas células exhiben un alto índice de proliferación y comparten varias similitudes fenotípicas con las células parentales de HTR8, incluyendo habilidades invasivas in vitro.
Las células se cultivaron a 37°C en atmósfera húmeda con 5% de CO2 en medio RPMI 1640 (Sigma Chemical Co.) suplementado con 10% de Suero Fetal Bovino (SFB) (Gibco®), 1% de L-Glutamina y 1% de PESTA (100U/ml de penicilina/ estreptomicina/ anfotericina). Las células fueron plaqueadas a platos de cultivo de 100 mm, incubadas por dos días en medio suplementado o hasta el 90% de confluencia y cultivadas durante 12 horas en medio libre de proteína, tras lo cual se colocaron los estímulos por el tiempo señalado.
Obtención del extracto total de proteínas
Para obtener el proteoma total, las células fueron lisadas empleando buffer RIPA (NaCl 150 mM, Tris-Cl pH 7,4 50 mM, EDTA 5 mM) con Tritón X-100 al 1% (Pierce), con coctel de inhibidores de proteasas y fosfatasas (Na3VO4 1 mM, PMSF 50 mM, Leupeptina 0,5 µg/µl, Pepstatina 0,5 µg/µl, Aprotinina 0,5 µg/µl); se incubó por 10 minutos a 4°C, tras lo cual se centrifugó a 10.000 rpm por 10 minutos, eliminando los restos celulares y conservando el extracto soluble.
Fraccionamiento celular
Se ensayó el fraccionamiento diferencial con detergentes descrito inicialmente por Rambsy y Makowski en 1999 (Walker, 2005), modificando los tiempos de incubación y los inhibidores de proteasas y fosfatasas empleados; y se implementó una serie de lavados tendientes no solo a limpiar el precipitado de cada fracción, sino también a aumentar la obtención de proteínas en la fracción soluble, al recuperar proteínas solubles ocluidas en el precipitado.
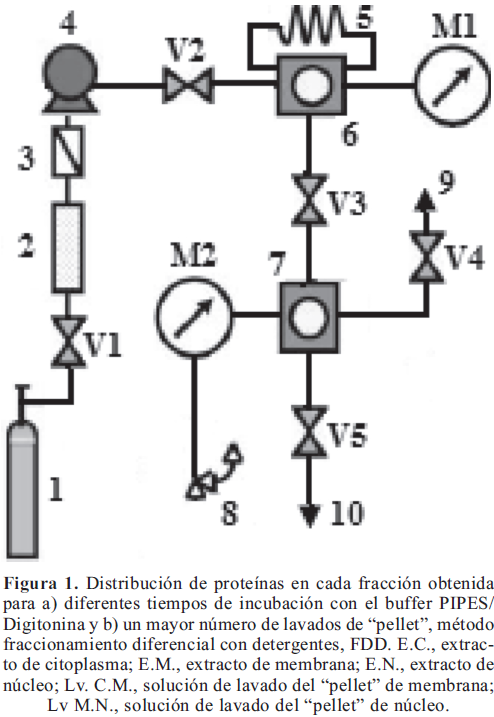
La fracción de citoplasma se obtuvo al permeabilizar la célula con el buffer PIPES (Sigma-Aldrich®)/Digitonina al 0,015%, con coctel de inhibidores de proteasas y fosfatasas (Na3VO4 1 mM, PMSF 50 mM, Leupeptina 0,5 µg/µl, Pepstatina 0,5 µg/µl, Aprotinina 0,5 µg/µl); se incubó por 10 minutos a 4°C, tras lo cual se centrifugó a 2.000 rpm por 10 minutos. El primer precipitado residual (C-M) que contiene los fantasmas celulares intactos fue lavado tres veces con 50 µl de buffer PIPES con coctel de inhibidores de proteasas y fosfatasas.
Para obtener la fracción de membrana, el precipitado fue solubilizado con el buffer PIPES (Sigma-Aldrich®) / Tritón X-100 (Merck®) al 0,5% con coctel de inhibidores de proteasas y fosfatasas (Na3VO4 1 mM, PMSF 50 mM, Leupeptina 0,5 µg/µl, Pepstatina 0,5 µg/µl, Aprotinina 0,5 µg/µl); se incubó por 30 minutos a 4°C, tras lo cual se centrifugó a 10.000 rpm por 10 minutos. Este segundo precipitado residual (M-N) que contiene las proteínas nucleares y las fibras del citoesqueleto fue lavado dos veces con 50 µl de buffer PIPES con coctel de inhibidores de proteasas y fosfatasas.
Cuantificación de proteínas
Cada extracto proteico obtenido se cuantificó por duplicado usando el kit de Pierce (Pierce®, Thermo scientific), diluyendo la muestra 20 veces y usando como estándar proteico BSA 2,0 – 0,0625 µg/µl. Se midió la absorbancia a 660 nm y se construyó la curva de calibración.
Electroforesis SDS-PAGE
Para observar las diferencias en los perfiles electroforéticos de cada fracción obtenida, 30 µg de proteína se separaron mediante SDS-PAGE en condiciones desnaturalizantes, con poliacrilamida al 10%. El gel obtenido se tiñó con Azul de Coomassie R-250 (al 0,1% en 3:6:1 MeOH : H2O : Ac. Acético) y fue enjuagado con agua destilada.
Western Blot
La presencia o ausencia de proteínas representativas de cada fracción se evaluó mediante Western Blot. 30 µg de proteína de cada fracción celular y lavado se separaron mediante SDS-PAGE al 10% en condiciones desnaturalizantes. Las proteínas se transfirieron a una membrana de nitrocelulosa; esta se bloqueó con leche al 5% en TBS (Tris-Base buffer Saline) a temperatura ambiente por dos horas y media, se incubó toda la noche a 4°C con el anticuerpo primario respectivo a una dilución de 1:1000 con leche al 1% en TBS, se realizaron 3 lavados de 10 minutos con TBS-Tween al 0,01%, se incubó con el anticuerpo secundario acoplado a peroxidasa de rábano a una dilución de 1:2000 para el anti-ratón o de 1:10.000 para el anti-conejo con leche al 1% en TBS. Se realizaron 3 lavados de 10 minutos con TBS-Tween al 0,01% y se reveló con luminol H2O2 a una película fotográfica fujifilm (Kodak®) por 5 minutos.
Los anticuerpos primarios empleados fueron inmunoglobulinas IgG policlonales de conejo anti-IGF-IR, anti- FAK, anti-ERK 1/2 y anti-Akt (todos de Santa Cruz Biotechnology, Inc.) e inmunoglobulinas IgG policlonales de ratón anti-p53 (Becton-Dickinson®). Los anticuerpos secundarios empleados fueron inmunoglobulinas IgG anticonejo y anti-ratón (Santa Cruz Biotechnology, Inc.)
Limpieza de sub-proteomas por precipitación
Se ensayaron dos métodos de precipitación para efectuar la limpieza de los proteomas, previo a su separación por isoelectroenfoque. En el método Ácido Tricloroacético/ Acetona, a la muestra de proteína se agrega ácido tricloroacético frío para una concentración final de 10% v/v y se incuba por 1 hora a -20°C, tras lo cual se centrífuga la muestra a 12.000 rpm, por 15 minutos a 4°C. Se lava dos veces con acetona fría y un último lavado con Acetona: Agua 90:10. Finalmente se disuelve en buffer de solubilización.
El método Cloroformo/Metanol se basa en una partición mediante la conformación de un sistema terciario Agua/Metanol:Cloroformo, en el que las proteínas permanecerán precipitadas en la interfase. En este 200 µl de extracto proteico se mezclan con 480 µl de MeOH y 160 µl de CHCl3 y luego con 640 µl H2O; se centrífuga por 5 minutos a 1.400 rpm, tras lo cual se succiona la capa superior y se adiciona 300 µl de MeOH. Se centrífuga por 30 minutos a 14.000 rpm a 4°C, y se deja secar al aire el precipitado de proteína. Este se re suspende en buffer de solubilización.
Separación de proteínas por electroforesis en dos dimensiones - 2D-PAGE
El extracto obtenido se precipitó con cloroformo/ metanol para concentrar la muestra y eliminar las sales, y se resuspendió en buffer de resolubilización (7 M urea, 2 M tiourea, 4% CHAPS, 40mM DTT, con anfolitos 0,5% en gradiente No Lineal 3-10).
Para la primera dimensión (Isoelectroenfoque), se empleó 700 µg de proteína en buffer de resolubilización; se llevó a 300 µl con buffer de rehidratación (7 M urea, 2 M tiourea, 2% CHAPS, 40 mM DTT, 1% anfolitos 3-10, y trazas de azul de bromofenol) y se usaron tiras secas inmovilizadas (18 cm, pH NL 4-7 de Bio-Rad®). La tira con la muestra se rehidrató de forma pasiva por 2 horas, y de forma activa por 11 horas a 50 V en un focalizador Bio- Rad®. El isoelectroenfoque de las tiras se llevó a cabo a los siguientes gradientes de voltaje: 250 V por 30’ (rampa rápida), 1.000 V por 30’ (rampa rápida), 1.000 V por 1h (constante), 4.000 V por 45’ (rampa rápida), 4.000 V por 1h (constante), y a 8.000 V (rampa rápida) hasta un total de 55 kVh.
Después del isoelectroenfoque, la tira fue incubada por 20 minutos en el primer buffer de equilibrio (5 ml, 6 M urea, 2% SDS, 0,375 M, pH 8.8, Tris-HCl, 20% glicerol, 130 mM DTT) e incubadas en el segundo buffer conteniendo iodoacetamida 135 mM en vez de DTT.
Para la electroforesis en dos dimensiones, las tiras IPG equilibradas fueron puestas en un gel de poliacrilamida al 12%. La SDS-PAGE fue llevada a cabo a 80 V por 12 horas y luego 90 V por 5 horas, usando una cámara de electroforesis DODECA Bio-Rad®. Como marcador de peso molecular se usó el estándar RPN5800 (GE®). Después de la separación, las proteínas en el gel fueron fijadas con etanol al 30% y ácido fosfórico al 2%; lavadas dos veces con ácido fosfórico al 2%; sensibilizadas en ácido fosfórico al 2%, etanol al 18% y sulfato de amonio al 15%; y teñidas con Coomassie coloidal (G-250) al 1%.
Resultados
Implementación del fraccionamiento celular
Para llevar a cabo el fraccionamiento celular se utilizó un fraccionamiento diferencial con detergentes usando un protocolo modificado desarrollado inicialmente por Rambsy y Makowski en 1999 (Walker 2005); este método permite obtener sub-proteomas gracias a la extracción secuencial con diferentes detergentes, aislando las proteínas de distintos compartimientos estructural y funcionalmente intactos. Inicialmente se ensayaron diferentes tiempos de incubación con el buffer PIPES/ Digitonina (5, 10, 15 y 20 minutos) y se midió la cantidad de proteína obtenida en cada extracto (Figura 1a). Ya que no se encontraron grandes diferencias en la cantidad de proteína de citoplasma obtenida, los análisis posteriores se llevaron a cabo a 5 y 10 minutos de incubación con el buffer PIPES/Digitonina. Si bien, se observó un contenido de proteína muy alto en la solución de lavado del primer precipitado (Lv C-M), el cual corresponde a los fantasmas celulares aún conteniendo la membrana citoplasmática, núcleo y citoesqueleto, y que debe ser lavado para terminar de retirar proteínas de citoplasma; esto implica la necesidad de implementar un mayor número de lavados de este primer precipitado (Figura 1b).
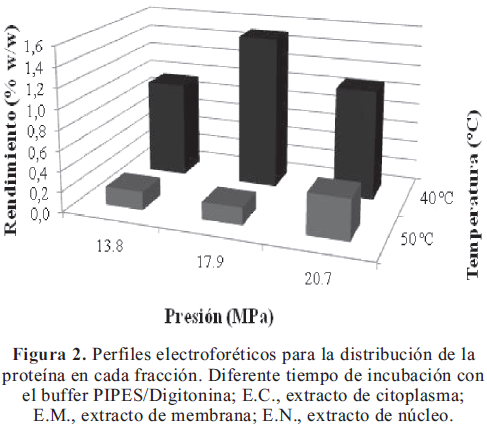
Mediante SDS-PAGE se comprobaron las diferencias en los perfiles electroforéticos de cada fracción, observando un fraccionamiento proteico al comparar cada extracto con el extracto total, con las mayores diferencias entre los perfiles del extracto de citoplasma y de membrana a un tiempo de incubación de 10 minutos (Figura 2).
Validación del fraccionamiento celular
Para evaluar el fraccionamiento celular se llevaron a cabo sendos Western Blot de proteínas representativas de cada fracción, tales como el receptor del factor de crecimiento similar a la insulina, IGF-IR, para membrana citoplasmática y la quinasa transductora de señal ERK para citoplasma. Los resultados mostraron que bajo las condiciones empleadas, fue posible obtener un extracto enriquecido en proteínas de citoplasma con muy baja contaminación con proteínas de membrana. Se encontró una muy baja contaminación (1,14%) por IGF-IR en el extracto de citoplasma después de diez (10) minutos de incubación con buffer digitonina, el cual no era detectable tras cinco (5) minutos de incubación. La solución de lavado también se encontró libre de proteínas de membrana, lo que indica que se mantiene la integridad celular en este punto (Figura 3a).
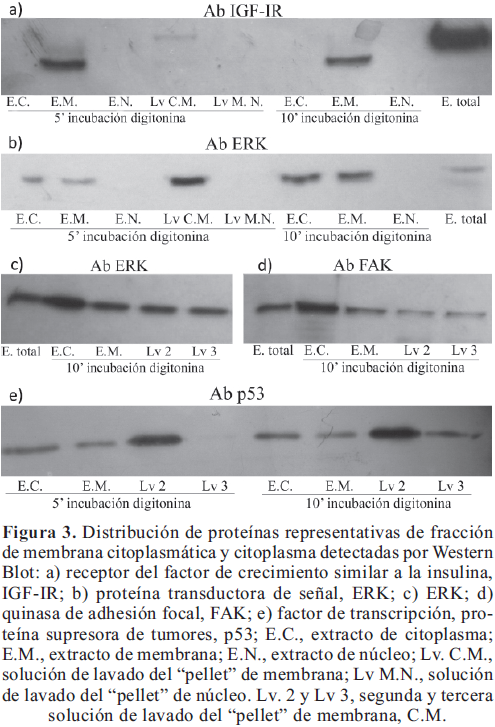
Sin embargo, al evaluar la distribución de la quinasa citoplasmática ERK (Figura 3b), se observó la presencia de ésta, tanto en el extracto citoplasmático como en el de membrana, y más abundantemente en la primera solución de lavado del primer precipitado (C-M). Este primer precipitado obtenido, que contiene los fantasmas celulares, se sometió a lavados con buffer PIPES para eliminar proteínas de citoplasma residuales al interior de la célula, encontrando un 88,59% de presencia de ERK en el extracto de membrana con respecto al extracto de citoplasma. De tal forma que el extracto de membrana se encontraba enriquecido en proteínas de membrana, pero contaminado con proteínas de citoplasma y era necesario implementar pasos adicionales de lavado del primer precipitado. Al incrementar el número de lavados del primer precipitado (C-M), se observa como disminuye la cantidad de proteína obtenida en la fracción de membrana, así como disminuye la cantidad de proteína presente en cada solución de lavado consecutiva (Figura 1b). No obstante al evaluar la distribución de proteínas citoplasmáticas como ERK (Figura 3c) y FAK (Figura 3d) en las distintas soluciones de lavado obtenidas, así como en la fracción de membrana, se sigue observando la presencia ubicua de estas proteínas. A pesar de lo anterior, el extracto de citoplasma mostró un enriquecimiento en ERK del 208,5% y en FAK del 185,6%, con respecto al extracto total (Figura 3c y 3d).
Si bien, hay que tener en cuenta que estas proteínas, a pesar de ser citoplasmáticas, frecuentemente se anclan a membrana donde llevan a cabo diferentes procesos de señalización: FAK, particularmente, forma complejos con integrinas y otras proteínas de membrana, vinculando también proteínas señalizadoras como ERK, de tal forma que se puede obtener tanto en el extracto de citoplasma como en el de membrana y la presencia de estas en el extracto de membrana no significa plenamente contaminación de este.
Para descartar que la presencia de proteínas de citoplasma en el extracto de membrana sea debida a la formación de estos complejos focales, se evaluó la presencia de una proteína que se mueve entre citoplasma y núcleo pero nunca llega a las inmediaciones de la membrana plasmática. Esta es el factor de transcripción p53, el cual se encontraba en las 2 fracciones y en los lavados, concluyendo que la fracción de membrana se encuentra contaminada con proteínas de citoplasma, a pesar de los lavados implementados (Figura 3e).
Finalmente, se obtuvo un extracto de citoplasma libre de proteínas de membrana y reproducible, mientras que el extracto de membrana mostró contaminación con proteínas de citoplasma, a pesar de las diferentes estrategias ensayadas.
Limpieza de la muestra y verificación de la reproducibilidad tras precipitación
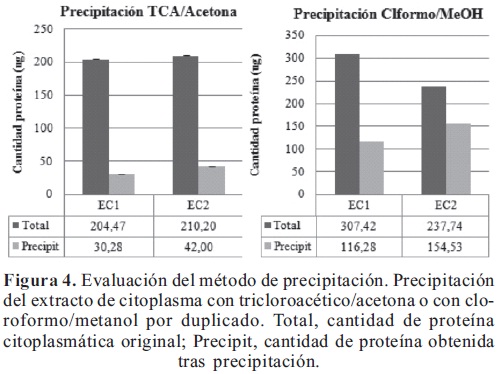
Para la evaluación de proteomas por 2D es necesario el cambio de composición del buffer en el que se encuentran las proteínas y la eliminación de sales y otros compuestos iónicos que puedan interferir con el isoelectroenfoque. Inicialmente se evaluó la precipitación con Ac. Tricloroacético / Acetona, obteniéndose una recuperación muy baja, a diferencia de la precipitación con Cloroformo / Metanol, la cual permite rendimientos y recuperaciones mayores (Figura 4).
Adicionalmente se evaluó la reproducibilidad del extracto de citoplasma obtenido y se verificó si hay pérdidas selectivas de proteína durante la precipitación; para esto se evaluaron por Western Blot la presencia de diferentes proteínas, como la quinasa transductora de señal, ERK, o la quinasa transductora de señal, Akt, en el extracto de citoplasma o inicial y en el extracto obtenido tras la precipitación con TCA/Acetona.
A pesar del bajo rendimiento registrado para la precipitación con Ac. Tricloroacético / Acetona, la reproducibilidad de la técnica es buena y no se registraron pérdidas selectivas de proteína durante la precipitación (Figura 5). De igual forma, para el caso del cloroformo se obtuvo una buena reproducibilidad tanto en los extractos de citoplasma como con los precipitados obtenidos, sin presentarse pérdidas selectivas de proteína, respecto a las proteínas evaluadas (Figura 6). De tal forma que es posible obtener extractos reproducibles, incluso a nivel del precipitado y sin pérdidas selectivas de material, aspecto muy importante a la hora de trabajar en proteómica.


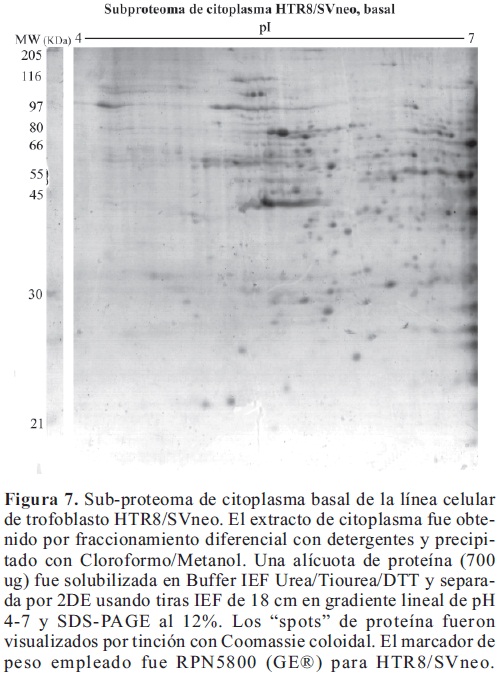
Obtención de geles bidimensionales
Para confirmar la limpieza de la muestra y su fraccionamiento, se analizó mediante electroforesis bidimensional el sub-proteoma de citoplasma obtenido con el fraccionamiento diferencial con detergentes, y limpiado y concentrado por precipitación con cloroformo/metanol. Se observó una buena separación y resolución de los "spots" tanto en la primera dimensión como en la segunda, así como una adecuada distribución de la muestra dentro del gel.
Al comparar el sub-proteoma de citoplasma obtenido con un perfil proteico proveniente del proteoma total (ver Figura 8), en general se observa un menor número de "spots" en el sub-proteoma, debido al fraccionamiento, siendo las proteínas de citoplasma el 46% del proteoma total; pero también se visualizan 85 "spots" que no tienen correspondencia con el proteoma total.

Los "spots" presentes en el sub-proteoma de citoplasma y ausentes en el proteoma total demuestran la importancia del fraccionamiento, ya que este, además de simplificar el análisis al disminuir la complejidad del extracto a analizar, permite mejorar la visualización de "spots" al enriquecerlos, que de otra forma quedarían ocultos debido al rango dinámico tan amplio de las proteínas.
Discusión
En general, cualquier manipulación aplicada a una mezcla compleja de proteínas va a conducir a algunas pérdidas inespecíficas de proteínas, como las que se registraron en la precipitación durante los pasos de preparación de la muestra. Este hecho sugiere que, cualquier pre-fraccionamiento de una muestra para análisis proteómico debe ser evitada. Sin embargo, la experiencia práctica reportada en diversos artículos y libros (Reiner Westermeier, 2008), demuestra que a pesar de lo anterior no existe ningún proceso que ofrezca un cubrimiento completo de un proteoma total y solo la pequeña combinación de técnicas y métodos permite resultados superiores. por lo tanto, la afirmación de que adquirir un proteoma total con solo un procedimiento es obsoleta, y no solo es viable, sino necesario realizar un fraccionamiento orientado a enriquecer las proteínas de interés, en este caso, proteínas de citoplasma y membrana. Este fraccionamiento celular es importante ya que gracias a él se logra reducir la complejidad de un proteoma, mejorando la separación y evitando la superposición de proteínas abundantes, y en consecuencia incrementando considerablemente el número de identificaciones.
por otro lado, el trabajo en proteómica requiere de alta reproducibilidad experimental que permita obtener datos biológicamente significativos y reproducibles. No obstante, para llevar a cabo esto es necesario implementar y validar las técnicas y métodos a emplear, asegurando la obtención de un sub-proteoma de citoplasma libre de proteínas de membrana y reproducible, para así poder determinar con certeza la ubicación celular de las proteínas a estudiar durante un análisis proteómico cualitativo, que pueda conducir a análisis proteómicos espacio-temporales.
Es posible determinar parcialmente la reproducibilidad de la técnica a nivel de los proteomas, obteniendo réplicas biológicas y técnicas; no obstante esto no permite determinar si hay pérdida selectiva de proteína durante la precipitación del extracto obtenido. En el presente caso, esto se logró al realizar Western Blot empleando anticuerpos específicos para diferentes proteínas, en los extractos de citoplasma antes y después de precipitar, siendo los geles de electroforesis 1-D y la técnica de Western Blot particularmente útiles para realizar una comparación preliminar de reproducibilidad de replicados de extractos.
Finalmente, el análisis de proteomas totales puede ser una labor ardua, por lo que es ideal realizar un fraccionamiento celular que permita disminuir la complejidad del extracto a analizar. Debido a que la línea de trofoblasto, HTR8/SVneo es un modelo interesante para comprender la patología del cáncer, el estudio de sus proteomas de membrana y de citoplasma es de gran interés.
En conclusión, la técnica de fraccionamiento con detergentes y precipitación con Cloroformo / Metanol empleada resultó adecuada para la obtención de un subproteoma de citoplasma de la línea celular de trofoblasto, HTR8/SVneo, con una reproducibilidad y recuperación adecuadas para su análisis por electroforesis bidimensional.
Financiación
Este trabajo fue financiado por la Dirección de Investigación de la sede Bogotá DIB, Universidad Nacional de Colombia (Proyectos Códigos: 10901 y 7193) y por Colciencias (Proyecto Código 110145221052).
Referencias bibliográficas
Dowling, P. M., P. Dowd, A. Henry, M. Glynn, S. Clynes, M. 2007. "Proteomic analysis of isolated membrane fractions from superinvasive cancer cells." Biochimica et Biophysica Acta 1774:93-101. [ Links ]
Everberg, H., T. Leiding, et al. 2006. "Efficient and non-denaturing membrane solubilization combined with enrichment of membrane protein complexes by detergent/polymer aqueous two-phase partitioning for proteome analysis." Journal of Chromatography A 1122(1-2): 35-46. [ Links ]
Ferretti, C., L. Bruni, et al. (2007). "Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts." Hum Reprod Update 13(2):121-141. [ Links ]
Foster, L. J., et al. 2005. "Differential expression profiling of membrane proteins by quantitative proteomics in a human mesenchymal stem cell line undergoing osteoblast differentiation". Stem Cells 23(9):1367-1377. [ Links ]
Gonnet, F., et al. 2003. "MALDI/MS peptide mass fingerprinting for proteome analysis: identification of hydrophobic proteins attached to eucaryote keratinocyte cytoplasmic membrane using different matrices in concert". Proteome Science 1(1):2. [ Links ]
Graham, C. H., T. S. Hawley, et al. 1993. "Establishment and Characterization of First Trimester Human Trophoblast Cells with Extended Lifespan". Experimental Cell Research 206(2):204-211. [ Links ]
Jethwaney, D., et al. (2007). "Proteomic analysis of plasma membrane and secretory vesicles from human neutrophils". Proteome Science 5(1):12. [ Links ]
Liang, X., et al. 2006. "Quantification of membrane and membranebound proteins in normal and malignant breast cancer cells isolated from the same patient with primary breast carcinoma." J. Proteome Res 5(10):2632-2641. [ Links ]
McCarthy, F. M., S. C. Burgess, et al. 2005. "Differential Detergent Fractionation for Non-electrophoretic Eukaryote Cell Proteomics." Journal of Proteome Research 4(2):316-324. [ Links ]
Payrastre, B., M. Plantavid, et al. 1988. "Characterization of plasma membranes from A431 cells, isolated by self-generating Percoll gradient: a rapid isolation procedure to obtain plasma membranes with functional epidermal growth factor receptors". Biochimica et Biophysica Acta (BBA) - Biomembranes 939(2):355-365. [ Links ]
Reiner Westermeier, T. N., Hans-Rudolf Höpker 2008. Proteomics in Practice: A Guide to Successful Experimental Design. Weinheim, Wiley-VCH. [ Links ]
Roobol-Bóza, M., V. Dolby, et al. 2004. "Membrane protein isolation by in situ solubilization, partitioning and affinity adsorption in aqueous two-phase systems: Purification of the human type 1 11[b]-hydroxysteroid dehydrogenase." Journal of Chromatography A 1043(2):217-223. [ Links ]
Selinsky, B. S. (Ed.) 2003. Membrane Protein Protocols: Expression, Purification, and Characterization. Methods in Molecular Biology, Humana Press. [ Links ]
Vertommen, A., B. Panis, et al. 2011. "Challenges and solutions for the identification of membrane proteins in non-model plants." Journal of Proteomics 74(8):1165-1181. [ Links ]
Walker, J. M., (Ed.) 2005. The Proteomics Protocols Handbook. Totowa, New Jersey, Humana Press Inc. [ Links ]
Zhang, L., et al. 2008. "Dataset of the plasma membrane proteome of nasopharyngeal carcinoma cell line HNE1 for uncovering protein function." Acta Biochimica et Biophysica Sinica 40(1):55-70. [ Links ]
Recibido: junio 20 de 2011. Aceptado para su publicación: agosto 30 de 2011
