










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Academia Colombiana de Ciencias Exactas, Físicas y Naturales
Print version ISSN 0370-3908
Rev. acad. colomb. cienc. exact. fis. nat. vol.35 no.136 Bogotá July/Sept. 2011
BÍOQUIMICA
1 Grupo de Investigación en Hormonas, Departamento de Química, Facultad de Ciencias, Universidad Nacional de Colombia, Bogotá.
2 Department of Clinical Sciences, Molecular and Translational Endocrinology Group, University of Las Palmas de Gran Canaria - Cancer Research Institute of The Canary Islands (ICIC), Spain.
* Correspondencia: email: mysanchezd@unal.edu.co mysanchezd@unal.edu.co
Abstract
Statins have been widely used for the treatment of hypercholesterolemia and other cardiovascular diseases. Recently, statins have been studied for their apoptotic effects which make them relevant for cancer prevention and treatment; however, their exact mechanisms of action are still unclear. In this study, we used malignant UMR-106 osteosarcoma cells and normal HTR8/SVneo extravillous trophoblast cells, and found that simvastatin decreases cell viability, in a dose and time-dependent manner in both cell types. In addition, 10 µM simvastatin was able to induce apoptosis in trophoblast cells as evaluated by FACS analysis. Finally, proteomic analysis of protein expression suggests a specific regulatory mechanism that could explain some of the anticancer effects of this statin.Key words: cancer, growth inhibition, statins, simvastatin, apoptosis.
Resumen
Las estatinas se han usado ampliamente en el tratamiento de la hipercolesterolemia y otras enfermedades cardiovasculares. Recientemente se han estudiado sus efectos apoptóticos lo que las hace relevantes en la prevención y tratamiento del cáncer, aunque sus mecanismos de acción se desconocen. En este estudio empleamos células malignas de osteosarcoma UMR-106 y células normales de trofoblasto extravelloso HTR8/SVneo y encontramos que la simvastatina, reduce la viabilidad celular de manera dependiente del tiempo y la dosis, en ambos tipos celulares. Además, mediante FACS se demostró la inducción de apoptosis por simvastatina 10 µM en células trofoblásticas. por último, el análisis proteómico sugiere un mecanismo regulatorio específico que podría ayudar a explicar los efectos anticancerosos de esta estatina.
Palabras clave: cáncer, inhibición crecimiento, estatinas, simvastatina
Introduction
Statins have been widely used for the treatment of hypercholesterolemia and other cardiovascular diseases. In the past few years, interest has grown around these drugs as an alternative for cancer treatment, as a response to the increasing amount of information about clinical and molecular evidence that statins in fact, can attack cancer cells, in a cell type-specific manner (Gauthaman, et al. 2009, Mueck, et al. 2003, Wong, et al. 2002). Statins decrease cholesterol synthesis by inhibiting the rate-limiting enzyme, 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase, involved in the mevalonate and cholesterol synthesis (Riganti, et al. 2008). The mechanism by which statins exert their effects on cells is not well known, although most biological effects have been widely documented (Aberg, et al. 2008, Koyuturk, et al. 2004, 2007). Considering the growing information obtained on statins, research is necessary to understand the targets of this drug and the potential role as adjuvant in therapy. In a previous study it was found that simvastatin, a lipophilic statin, has wide effects on the osteoblast-like osteosarcoma cell line UMR- 106 (Cabrera-Benitez, 2008). Osteosarcoma is the second cancer-related cause of death among children and young adults resulting in tumors on tibia, femur and humerus (Heare, et al. 2009). The aim of the present study was to investigate the effects of simvastatin on cell viability and apoptosis, using two different cell lines: osteosarcoma cell line UMR-106 and trophoblast cell line HTR8/SVneo, the later of interest considering its high proliferation and invasion levels similar to cancer cells. A preliminary analysis of the protein expression associated with simvastatin treatment in osteosarcoma cells was examined using a proteomic approach.
Materials and methods
Cell Culture
UMR-106, rat osteosarcoma cells and HTR8/SVneo, human trophoblast cells, were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 10% FBS, 2 mM glutamine, 100 Units/ml penicillin and 100 µg/ml streptomycin. Cells were maintained at 37ºC in a humidified atmosphere with 5% carbon dioxide. For simvastatin treatment, cell media was refreshed and simvastatin or vehicle was added in the indicated concentration.
Cell Viability Assay
Cell viability was assayed by measuring the mitochondrial reduction of the tetrazolium salt ([3-(4,5-dimethylthiazol- 2yl-)-2,5diphenyltetrazolium bromide], MTT) (Carmichael, et al. 1987). Cells were seeded in 96-well plates in medium supplemented with 10% FBS at a cell density of 20000 cells/well. Twenty-four hours later, simvastatin was added to the medium at the indicated concentrations (0.03 to 10 µM) or vehicle (0.01% ethanol) and cells were cultured for the indicated times. MTT (0.5 mg/ml) was added to each well for the last four hours and incubated at 37ºC. The medium was then discarded and the formazan precipitate was solubilized by addition of 20% SDS in 0.02N HCl for 12-16 hours. The optical density was measured at 595 nm with an ELX 800 Universal Microplate reader (Bio-TEK Instruments, INC.). Data was expressed as percent growth above the level in controls and the results in figures were plotted as means ± SEM of each test point from 3 separate experiments. IC50 was determined from a plot when fitted to a sigmoidal dose-response curve.
Flow cytometry
Detection and quantification of apoptosis was done by flow cytometry, double staining with Annexin V-FITC (AbD Serotec) and Propidium Iodide (PI). HTR8/SVneo cells were plated in 60 mm dishes to confluence, FBS deprived for 8 hours and simvastatin 1 or 10 µM was added for 2 or 48 hours. As a positive control of apoptosis cells were treated with 3% formaldehyde for 30 minutes. Cells were tripsinized for 5 minutes and collected by centrifugation. The pellet was washed with 1% Bovine Serum Albumin (BSA). Afterwards, 106 cells were labeled with 1 µg Annexin V-FITC for 10 minutes and 1 µg/mL PI, using Annexin V FITC kit (AbD Serotec, Oxford, UK), according to manufacturer’s instructions. Flow cytometry analysis was performed on a FACScan using CellQuest Pro software (BD Biosciences, Mountain View, CA) analyzing 10000 events for each treatment.Preparation of whole cell extract
UMR-106 cells were grown to confluence in 100 mm cell culture dishes and incubated as mentioned above. Cells were treated with 10 µM simvastatin or vehicle for 8 hours in fresh DMEM supplemented with 10% FBS. Cells were rinsed with ice-cold PBS, scrapped with lysis buffer (RIPA buffer, 10 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 2 mM phenylmethylsulfonyl fluoride (PMSF), 10 mM Na3VO4, 2 µg/mL aprotinin, 2 µg/mL leupeptin, 2 µg/mL pepstatin) and incubated on ice for 15 min. Lysates were centrifuged and supernatants were used as whole cell extract. Protein concentration was determined by the Bradford method (Bradford, 1976).Phenol extraction
Prior to 2D electrophoresis, proteins were extracted from whole cell extracts using pH 8.0 Tris-HCl-saturated phenol, in order to remove interferences such as salts, lipids and nucleic acids as reported previously (Arévalo-Ferro, 2004). Briefly, 1mL aliquots from the protein extracts were mixed by vortexing with 1mL phenol and incubated for 10 min at 70°C. The sample was cooled on ice for 5 min and the phases were separated by centrifugation at 5000 rpm for 10 min. at 4°C. The aqueous phase was discarded and 1mL of distilled water was added. This procedure was repeated once and proteins were precipitated with 1 mL of ice-cold acetone. The pellet was recovered by centrifugation at 15000 rpm for 20 min 4°C and washed with 1 mL of acetone, centrifuging for 10 min at 4°C 15000 rpm. After the pellet was dried, proteins were resuspended in 300 µL of solubilization buffer (8M Urea, 2% CHAPS, 0.2% Carrier ampholytes pH 3-10, 2.8 mg/mL 1,4-dithiothreitol (DTT), and a trace of bromophenol blue).Two-dimensional Electrophoresis(2-DE)
Two-dimensional electrophoresis was carried out according to the manufacturer’s instructions. Briefly, protein samples were resuspended in solubilization buffer, as described above and 18 cm pH 3-10 nonlinear strips were used (Bio-Rad®). Strips were passively rehydrated with sample containing 500 µg protein for 2 hours, followed by active rehydration at 50 V for 11 hours performed in the Protean IEF System (Bio-Rad®, USA) First-dimension isoelectric focusing (IEF) was carried out increasing linearly from 250V to 8000 V until reaching 55kVh. The IPG strip was then equilibrated in 6 M urea, 30% w/v glycerol, 2% w/v SDS, 0.05 M Tris-HCl pH 8.8, 2% w/v DTT and then with 2.5% w/v iodoacetamide. Second dimension, SDS-PAGE was carried out on a 12% polyacrylamide gel in the Bio-Rad vertical system (230 × 200 × 1.5 mm; Gibco®, USA) at a constant voltage of 80V for 12 hours and 90 V for 5 hours. Gels were visualized using Colloidal Coomassie staining and then analyzed qualitatively and quantitatively by PDQuest Software (Bio-Rad®). Spot intensities were analyzed using pixel units and compared between the two gels.
Results
Simvastatin reduces cell viability of osteosarcoma and trophoblast cells
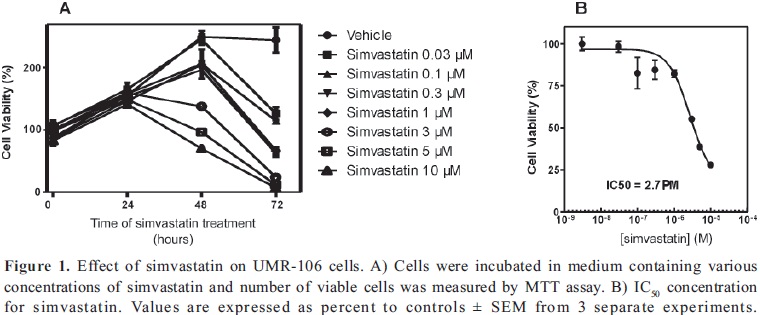
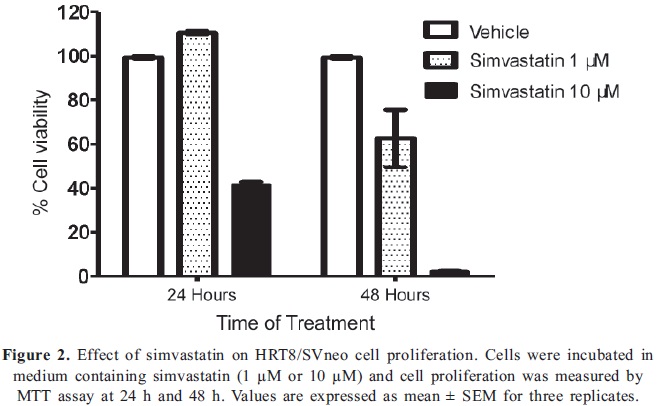
First, we examined the effect of simvastatin on UMR- 106 cell viability by MTT assay. The viable cell number of UMR-106 was decreased in a dose and time dependent manner (Fig 1A). We found that doses of simvastatin higher than 1 µM significantly reduced cell viability up to 72% when cells were treated with 10 µM simvastatin compared to values of control cells, after 48 h of exposure to the drug. IC50 value was measured to be 2.7 µM simvastatin, concentration needed to reduce growth of osteosarcoma cells by 50%, (Fig 1B). To compare the effect of the statin on the growth of a normal cell line, we used the trophoblast HTR8/SVneo cell line. Cells were not affected by 1µM simvastatin at 24 hours, however, doses as high as 10 µM decreased cell viability by 60% at 24 h and had a profound cytotoxic effect at 48 h, compared to control cells (Fig 2).
Apoptosis in HTR8 cells is increased by 10 µM simvastatin
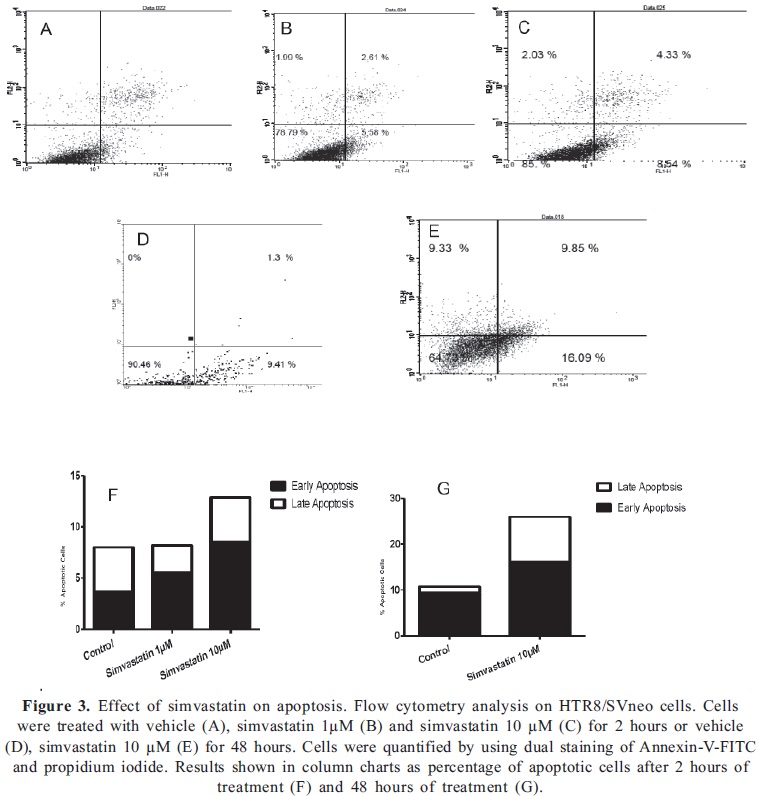
We determined whether simvastatin could induce apoptosis in HTR8/SVneo cells. As shown in Fig. 3, simvastatin induced a dose-dependent early apoptotic effect after 2 hours of treatment. The effect was more notable with 10 µM simvastatin as observed with the increase in apoptotic cells form 8.8% to 12.8% (Fig 3a). After 48 hours, the effect was intensified and we observed a raise in apoptotic cells from 10.7% to 25.9% compared to cells treated with vehicle. Both early and late apoptotic cells were increased after simvastatin treatment, indicating that this statin can influence cell viability and proliferation in trophoblast cells by the induction of apoptosis.
Proteomic profile of UMR-106 cells is influenced by simvastatin treatment
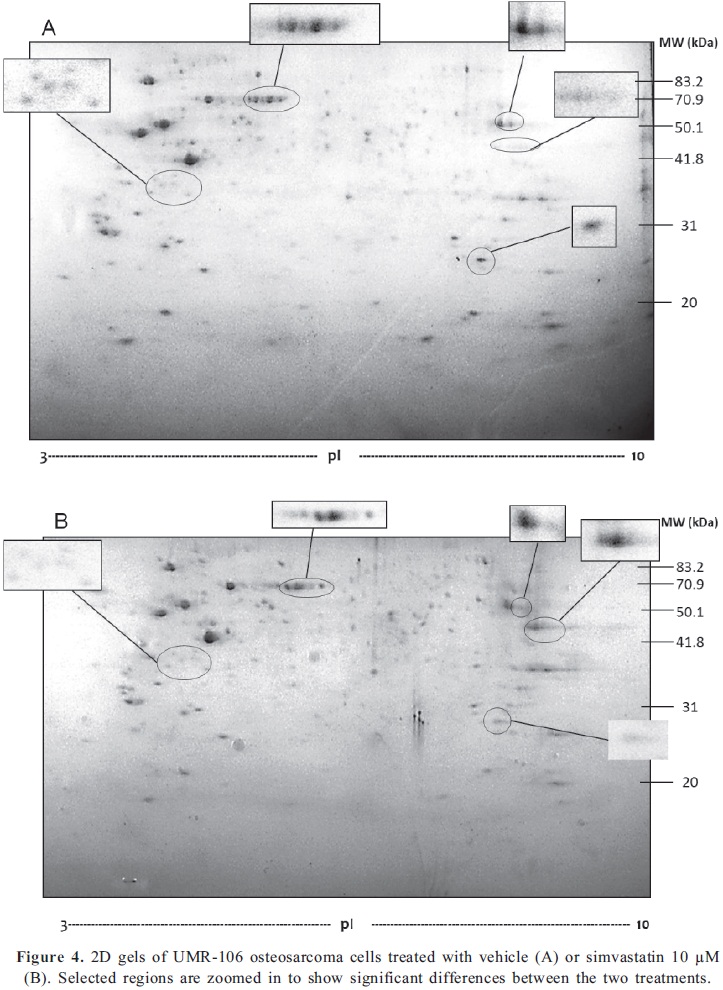
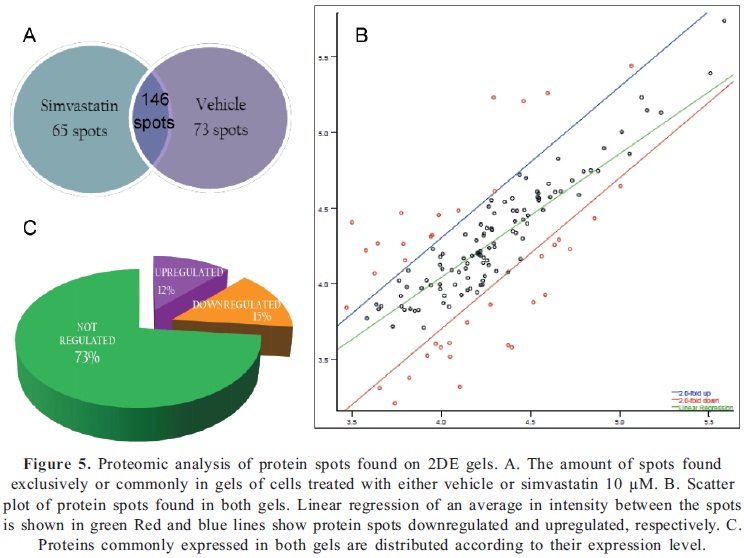
The molecular mechanisms by which statins exert their effects on cells are poorly documented. We investigated the changes in global protein expression by two-dimensional electrophoresis (2-DE) of cells exposed to 10µM simvastatin for 48 h, in comparison with cells treated with vehicle. The 2-DE gel images obtained are shown in Fig 4A and B and some of the spots that were found to be differentially expressed between the two conditions are highlighted. This illustrates some of the most noticeable differences in expression comparing both gels. Gel images were analyzed using PD Quest Software and results revealed that 211 protein spots were found in cells treated with simvastatin, compared to 219 protein spots found in control gel. Through densitometry analysis we were able to determine that a total of 146 protein spots were found to be common to both conditions, whereas 65 spots were only expressed in cells under statin treatment and 73 spots were exclusively found in control cells (Fig 5A). A scatter plot illustrates the regulation profile of matched proteins in both gels, where 18 spots (12%) were at least 2 fold up-regulated and 21 spots (15%) were at least 2 fold down-regulated by simvastatin, compared to the control cells (Fig. 5B). To summarize, Fig 5C shows the distribution of matched proteins.
Discussion
Simvastatin is a lipophilic statin, highly used in the treatment of cardiovascular diseases. Like other statins, inhibits the rate-limiting step of cholesterol biosynthesis, the conversion of 3-hydroxy-3-methylglutarylCoA (HMGCoA) to L- mevalonic acid, through binding to HMG-CoA reductase’s active site. In addition to a decrease in hepatic cholesterol synthesis, statins could also inhibit the synthesis of important isoprenoid intermediates, such as dolychol phosphate, farnesylpyrophosphate (FPP) and geranylgeranylpyrophosphate (GGPP). These intermediates have a role in N-linked glycosylation and as lipid attachments for membrane proteins involved in signal transduction (Casey, 1992, Sinensky, 2000).
Several studies worldwide have shown the effect of simvastatin and other statins in cell viability, migration and invasion levels (Hoque, et al. 2008, Kidera, et al. 2010, Koyuturk, et al. 2004, Wong, et al. 2002). Some studies have shown a decreased risk of developing cancer in general in patients that regularly take statins as hypercholesterolemia therapy (Demierre, et al. 2005, Stamm, et al. 2005). However, the exact mechanisms of their anticancer action are still unclear. In this study, we found that simvastatin decreases cell viability, in two different cell types, in a dose and time-dependent manner. UMR-106 cells, derived from rat osteosarcoma, were significantly affected by simvastatin, as deduced by the marked reduction in cell viability and the relatively low IC50 value for the drug. The IC50 concentration, which is indicative of the levels of cytotoxicity of simvastatin, varies according to the cell type in a wide range (Guerrero, et al. 2010). The IC50 value of 2.7ìM we found for osteosarcoma cells is lower than those reported for simvastatin and other statins structures (Gronich, et al. 2004, Guerrero, et al. 2010, Kang, et al. 2005, Koyuturk, et al. 2007). In a review by Graaf et al.(2004) is stated that a mutation on p53 protein might increase the IC50 value found in several cancer cell lines for statins and farnesyl transferase inhibitors. Therefore, UMR-106 osteosarcoma cell line is particularly sensitive to simvastatin compared with other cancer cells. To our knowledge, no p53 mutations have been reported on this osteosarcoma cell line so far and it was previously reported that the simvastatin-induced cell cycle arrest is p53 independent (Cabrera-Benitez, 2008).
Inhibitory effects of statins have also been examined in normal tissues. Although the placenta is considered a normal tissue, its constitutive trophoblastic cells are characterized by high rates of proliferation and invasion, similar to the behavior of cancer cells. These abilities make them ideal for monitoring molecular changes taking place during the acquisition of a malignant/invasive phenotype. In previous studies, placental tissues treated with simvastatin revealed similar results as those obtained with malignant tissues. When explants are treated with doses of the statin up to 10 µM, a significant decrease in cell proliferation and an increase in apoptosis events were shown (Kenis, et al. 2005). In the present study, we used an immortalized extravillous cell line, HTR-8/SVneo, derived from trophoblast cells of the first trimester of pregnancy, where growth plays a critical role in the development of the fetus and could demonstrate the growth inhibition and pro-apoptotic effects of simvastatin as well.
Statins effects on cell signaling are not completely understood, but it seems to be a pleiotropic effect, and it does not only compromise a particular pathway, but rather a whole network involving the cholesterol biosynthesis and its precursors. Cholesterol has been found to have an essential part in the growth of all eukaryotic cells, especially in rapidly proliferating cells, which usually have high levels of cholesterol synthesis and HMG-CoA reductase (Siperstein, 1984). Mevalonate biosynthesis catalyzed by HMG-CoA reductase is the starting step for a chain reaction that involves the biosynthesis of isoprenes, which are implicated in DNA synthesis initiation (Larsson, 1996). Thus, decreasing HMG-CoA reductase activity can also regulate cell growth and proliferation, as proven with our results. Nevertheless, the role of statins as inhibitors of farnesylated and geranylgeranylated proteins cannot be discarded. Statin treatment also inhibits isoprene synthesis, which directly affects posttranslational modifications necessary for proteins such as Rho, Ras, Rac and Cdc42, to anchor to the membrane, and all the conformational changes needed for the signaling pathway to start. Proteins from the Ras superfamily are involved in several biological processes including proliferation, migration and invasion of both cancerous and non-cancerous cells, therefore, a downregulation on either of these pathways might also lead to decreasing proliferation and apoptosis. Further studies carried out in our research group point towards the participation of other signaling pathways also involved in cell malignization like JAK/STAT pathway and its negative regulators, SOCS proteins. We found SOCS-3 and CIS mRNA levels to be upregulated with simvastatin treatment (Sandoval-Usme, et al. 2010). Additional studies are required to investigate which are the molecular effects of statins in signaling pathways and how SOCS proteins and JAK/STAT pathway might be implicated in simvastatin effects on osteosarcoma cells.
Finally, in order to get insight into the potential targets of simvastatin action, we examined the global changes in protein profile of statin treated cells. Using a proteomic approach we found that simvastatin exposure results in changes in the cellular protein profile, regulating the level of expression of already expressed proteins and inducing the expression of new ones. All this lead us to think that simvastatin effects on protein expression are very specific, probably depending more likely on those exclusively expressed proteins, than on the regulation of those preexisting proteins. However, regulated proteins cannot be discarded, considering that changes in expression were significant and they might also represent important changes in signaling pathways, which would allow us to further clarify the effects of statin on cancerous and noncancerous cells. Protein identification was out of the scope of this paper, but future mass spectrometry studies can give us a broader vision of the differences in protein signaling and also provide us with other protein targets.
Conclusions
Simvastatin induces apoptosis and inhibits proliferation in a time and dose-dependent manner in normal and cancerous cells. Proteomic analysis of protein expression gives support to a specific regulatory mechanism that could explain some of the anticancer effects of this statin. Further research is needed to identify the proteins responsible for this action.
Aknowledgments
This work has been supported by grants from the Dirección de Investigación Bogotá, DIB, Universidad Nacional de Colombia (Projects: 10878 and 12243) and Colciencias (Project: 110145221052).
HTR8/Svneo cell line was kindly donated by Dr. Angela Cadavid from Universidad de Antioquia, Medellín, Colombia.
References
Aberg, M., Wickstrom, M. and Siegbahn, A. 2008. Simvastatin induces apoptosis in human breast cancer cells in a NFkappaBdependent manner and abolishes the anti-apoptotic signaling of TF/FVIIa and TF/FVIIa/FXa, Thromb Res 122(2):191-202. [ Links ]
Arévalo-Ferro, C. 2004. Proteomics View Of Quorum-Sensing Regulated and Surface Induced Genes in Representative Pseudomonas and Burkholderia Species., 128, PhD, Institute of Microbiology of Technischen München, Germany. [ Links ]
Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal Biochem 72:248-254. [ Links ]
Cabrera-Benitez, N. E. 2008. Papel de los inhibidores de la 3- hidroxi-3-metilglutaril coenzima A reductasa (estatinas) en las líneas celulares de osteosarcoma. Estudio In vitro., 141, PhD, Departamento de Ciencias Clínicas, Las Palmas de Gran Canaria, Gran Canaria, Spain. [ Links ]
Carmichael, J., DeGraff, W. G., Gazdar, A. F., Minna, J. D. and Mitchell, J. B. 1987. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing, Cancer Res 47(4):936-942. [ Links ]
Casey, P. J. 1992. Biochemistry of protein prenylation, J Lipid Res 33(12):1731-1740. [ Links ]
Demierre, M. F., Higgins, P. D., Gruber, S. B., Hawk, E. and Lippman, S. M. 2005. Statins and cancer prevention. Nat Rev Cancer 5(12):930-942. [ Links ]
Gauthaman, K., Manasi, N. and Bongso, A. 2009. Statins inhibit the growth of variant human embryonic stem cells and cancer cells in vitro but not normal human embryonic stem cells. Br J Pharmacol 157(6):962-973. [ Links ]
Gronich, N., Drucker, L., Shapiro, H., Radnay, J., Yarkoni, S. and Lishner, M., 2004. Simvastatin induces death of multiple myeloma cell lines, J Investig Med 52(5):335-344. [ Links ]
Guerrero, M., Di Giulio, C. and De Sanctis, J. B. 2010. Las estatinas afectan la viabilidad de líneas celulares de leucemia y linfoma humanas in vitro, Revista de la Facultad de Medicina 33(1):11-14. [ Links ]
Heare, T., Hensley, M. A. and Dell'Orfano, S. 2009. Bone tumors: osteosarcoma and Ewing's sarcoma, Curr Opin Pediatr 21(3):365-372. [ Links ]
Hoque, A., Chen, H. and Xu, X. C. 2008. Statin induces apoptosis and cell growth arrest in prostate cancer cells, Cancer Epidemiol Biomarkers Prev 17(1):88-94. [ Links ]
Kang, W. K., Lee, I., Ko, U. and Park, C. 2005. Differential effects of RhoA signaling on anticancer agent-induced cell death, Oncol Rep 13(2):299-304. [ Links ]
Kenis, I., Tartakover-Matalon, S., Cherepnin, N., Drucker, L., Fishman, A., pomeranz, M. and Lishner, M. 2005. Simvastatin has deleterious effects on human first trimester placental explants, Hum Reprod 20(10):2866-2872. [ Links ]
Kidera, Y., Tsubaki, M., Yamazoe, Y., Shoji, K., Nakamura, H., Ogaki, M., Satou, T., Itoh, T., Isozaki, M., Kaneko, J., Tanimori, Y., Yanae, M. and Nishida, S. 2010. Reduction of lung metastasis, cell invasion, and adhesion in mouse melanoma by statin-induced blockade of the Rho/Rhoassociated coiled-coil-containing protein kinase pathway, J Exp Clin Cancer Res 29: 127. [ Links ]
Koyuturk, M., Ersoz, M. and Altiok, N. 2004. Simvastatin induces proliferation inhibition and apoptosis in C6 glioma cells via cjun N-terminal kinase, Neurosci Lett 370(2-3):212-217. [ Links ]
Koyuturk, M., Ersoz, M. and Altiok, N. 2007. Simvastatin induces apoptosis in human breast cancer cells: p53 and estrogen receptor independent pathway requiring signalling through JNK, Cancer Lett 250(2):220-228. [ Links ]
Larsson, O. 1996. HMG-CoA reductase inhibitors: role in normal and malignant cells, Crit Rev Oncol Hematol 22(3):197-212. [ Links ]
Mueck, A. O., Seeger, H. and Wallwiener, D. 2003. Effect of statins combined with estradiol on the proliferation of human receptor-positive and receptor-negative breast cancer cells, Menopause 10(4):332-336. [ Links ]
Riganti, C., Aldieri, E., Doublier, S., Bosia, A. and Ghigo, D. 2008. Statins-mediated inhibition of rho GTPases as a potential tool in anti-tumor therapy, Mini Rev Med Chem 8(6):609-618. [ Links ]
Sandoval-Usme, M. C., Umaña-Pérez, A., García-Castellano, J. M., Fernández-Pérez, L. F. and Sánchez-Gómez, M. 2010. OR5,28 GH-induced activation of JAK/STAT signaling in osteosarcoma cells is inhibited by statins through the induction of SOCS expression, Growth Hormone and IGF Research, 20(1 (supplemented))S13-S13. [ Links ]
Sinensky, M. 2000. Recent advances in the study of prenylated proteins, Biochim Biophys Acta 1484(2-3):93-106. [ Links ]
Siperstein, M. D. 1984. Role of cholesterogenesis and isoprenoid synthesis in DNA replication and cell growth, J Lipid Res 25(13)1462-1468. [ Links ]
Stamm, J. A. and Ornstein, D. L. 2005. The role of statins in cancer prevention and treatment, Oncology (Williston Park), 19(6):739-750; discussion 753-734. [ Links ]
Wong, W. W., Dimitroulakos, J., Minden, M. D. and Penn, L. Z. 2002. HMG-CoA reductase inhibitors and the malignant cell: the statin family of drugs as triggers of tumor-specific apoptosis, Leukemia 16(4)508-519. [ Links ]
