










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Academia Colombiana de Ciencias Exactas, Físicas y Naturales
Print version ISSN 0370-3908
Rev. acad. colomb. cienc. exact. fis. nat. vol.35 no.136 Bogotá July/Sept. 2011
QUÍMICA ANALÍTICA
Mónica Cala, Ángela Vásquez, Alejandro García , Jairo René Martínez, Elena Stashenko1
Resumen
Se implementaron metodologías de análisis por electroforesis capilar (CE) y cromatografía líquida de alta eficiencia (HPLC) para la separación y cuantificación de (±)-catequina, (-)- epicatequina, (-)-epigalocatequina, galato de (-)-epicatequina y galato de (-)-epigalocatequina, en 5 muestras de 3 variedades de cacao cultivadas en Colombia. La técnica de HPLC fue más reproducible que la CE, que sin embargo, resultó ser más rápida, económica y menos contaminante. Se cuantificaron, por HPLC y CE (estándar externo), (±)-catequina (0,07-0,79 mg/g de cacao) y (-)- epicatequina (1,99-9,66 mg/g de cacao) en los extractos acuosos de las tres variedades de cacao.
Palabras clave: catequinas, CE, HPLC, Theobroma cacao.
Abstract
Analytical methodologies based on capillary electrophoresis (CE) and high performance liquid chromatography (HPLC) were implemented for the separation and quantitation of (±)-catechine, (-)-epicatechine, (-)-epigallocatechine, (-)-epicatechine gallate and (-)-epigallocatechine gallate in 5 samples of 3 cacao varieties grown in Colombia. HPLC was found to be more reproducible than CE, which turned out to be faster, more economical and less contaminant. (±)-Catechine (0,07- 0,79 mg /g cacao) and (-)-epicatechine (1,99-9,66 mg /g cacao) were determined in aqueous extracts of the 3 cacao varieties.
Key words: catechines, CE, HPLC, Theobroma cacao.
Introducción
El gran interés en el consumo de productos como chocolate y té, en los hábitos alimenticios de diferentes países, se atribuye a su contenido de compuestos fenólicos (Folch-Cano, C. et al. 2010; Yilmaz, 2006), entre los cuales se destacan (±)-catequina [(±)-C], (-)-epicatequina [(-)-EC], (-)-epigalocatequina [(-)-EGC], galato de (-)-epicatequina [(-)-ECG] y galato de (-)-epigalocatequina [(-)-EGCG], por su efecto antioxidante, ampliamente estudiado (Tsao y Deng, 2004; Putte y Hollman, 2000; Rice-Evans, 1996).
La implementación de metodologías que permitan la determinación y cuantificación de catequinas en muestras vegetales, es de importancia para tareas de bioprospección, desarrollo de ingredientes para las industrias farmacéutica, de alimentos y cosmética, para el control de calidad, y muchas otras aplicaciones. El desarrollo de técnicas instrumentales, que logren de manera rápida y eficaz la determinación de compuestos polares y no-volátiles, tales como la electroforesis capilar y la cromatografía líquida de alta eficiencia, ha resultado de gran utilidad para la identificación y cuantificación rápida de diversos tipos de catequinas, en matrices con diferentes características (Castro, M. et al. 2011; Calderón, A. et al. 2009). Aunque ambas técnicas han mostrado excelentes resultados en cuanto a la separación de catequinas, la discusión sobre cuál de las dos resulta más adecuada continúa abierta, ya que cada una presenta diferentes ventajas y desventajas, tal como lo muestran algunas publicaciones sobre el estudio comparativo de estas técnicas analíticas (Valls, J. et al. 2009; Bowser, M. et al. 2010; Dalluge y Nelson, 2000).
En el presente trabajo, se implementaron metodologías para la cuantificación de catequinas en cacao (variedades ICS-39, TSH-565 e IMC-67), basadas en las técnicas analíticas de cromatografía líquida de alta eficiencia (HPLC) y electroforesis capilar (CE).
Parte experimental
Material vegetal. Una muestra de cacao ICS-39, 2 muestras de la variedad TSH-565 y 2 muestras de la variedad IMC-67 fueron colectadas en la vereda El Rincón, a 6 km, sobre la vía que comunica con el municipio del Socorro (Santander). Las semillas de cada variedad fueron retiradas del fruto y posteriormente despulpadas. Luego de tres lavados sucesivos, se secaron a 30°C durante una semana hasta alcanzar peso constante. por último, las semillas se maceraron y se almacenaron a temperatura ambiente en recipientes plásticos rotulados.
Material de referencia y reactivos. El material de referencia certificado de las catequinas (±)-C (99%), (-)-EC (99%), (-)-EGC (99%), (-)-ECG (98%) y (-)-EGCG (95%), empleadas para la calibración, fue suministrado por Sigma- Aldrich (St. Louis, MO, EE.UU.). Agua, metanol, acetonitrilo grado HPLC, ácido clorhídrico (37% p/v) y cloroformo fueron adquiridos de J.T Baker (México D.C., México); ácido acético (98%) y acetato de etilo (99,8%) fueron obtenidos de Riedel-de Haën (Seelze, Suiza).
Preparación de las soluciones estándar. La solución madre (500 ppm), se preparó disolviendo (±)-C (0,50 mg), (- )- EC (0,50 mg), (-)-EGC (0,50 mg), (-)-ECG (0,50 mg) y (-)- EGCG (0,50 mg) en agua grado HPLC (1,0 mL). Las soluciones de trabajo para la curva de calibración se prepararon a partir de la solución madre, en el rango de 0,5 a 64 ppm.
Preparación de la muestra. Semillas de cacao en polvo (100 mg) se trataron con hexano (10 mL) y se centrifugación (3000 rpm/10 min). Se retiró el hexano y se secó la muestra con corriente de nitrógeno. Seguidamente, se agregó agua grado HPLC (10 mL), y se agitó la suspensión con ultrasonidos (15 min). posteriormente, la mezcla se centrifugó a 3000 rpm durante 20 min. El extracto acuoso obtenido, fue sometido a dos extracciones líquido – líquido sucesivas, primero, con cloroformo (10 mL) y después, con acetato de etilo (10 mL). De la primera extracción se tomó la fase acuosa y de la segunda extracción, se conservó la fracción orgánica. Finalmente, esta fracción se llevó a sequedad con corriente de nitrógeno y, el extracto después de pesado, se disolvió en agua grado HPLC y se aforó a 1,0 mL para su posterior análisis.
Análisis cromatográfico. El análisis cromatográfico se realizó usando un cromatógrafo líquido Agilent Technologies 1200 Series (Miami, FL, EE.UU.), que consta de un inyector manual Agilent Technologies 1200 G1328B; una bomba cuaternaria, Agilent Technologies G1354A; un detector UV-Vis de arreglo de diodos (DAD), Agilent Technologies G1315B, usado para determinar la pureza del pico cromatográfico. La identificación e integración de picos se realizó a través del software Agilent ChemStation (G1701-DA). La separación cromatográfica se realizó con un guarda-columna Agilent Technologies ECLIPSE XDBC18 (4,6 x 12,5 mm, 5 µm) y una columna apolar, ZORBAX Eclipse XDB-C18 (4,6 x 150 mm, 5 µm). La temperatura de la columna se mantuvo en 35°C. La detección de las cinco catequinas fue realizada a una longitud de onda de 210 nm. La fase móvil seleccionada fue una mezcla de dos solventes, (A) metanol y (B) agua:ácido acético (100:0,1). La cromatografía isocrática se desarrolló con flujo de 1 mL/ min y una relación A:B (18:82).
Para el análisis por electroforesis capilar se empleó un equipo Lumex modelo CAPEL 105 (San Petersburgo, Rusia), con detector UV (190 – 380 nm), un capilar de sílice fundida de 75 µm i.d., con largo total y efectivo de 60 y 50.5 cm, respectivamente, y una fuente de poder con polaridad positiva. El tratamiento de los datos obtenidos utilizó el software Chromatography Data Station, Chrom&Spec, Versión 1.5. La solución buffer empleada fue una mezcla de ácido bórico (35 mM), dihidrógeno fosfato de sodio (20 mM) y dodecil sulfato de sodio (80 mM). La separación electroforética se realizó a 20 kV y 20°C durante 14 min. La detección se realizó a 210 nm. Para el acondicionamiento diario del capilar, al inicio de cada jornada, se realizó un lavado con agua (3 min), metanol (3 min), ácido clorhídrico (5 min) e hidróxido de sodio (15 min) y para el lavado entre corridas se empleó la misma solución buffer descrita anteriormente (5 min).
Implementación de los métodos de análisis. Para el análisis simultáneo por HPLC de (±)-C, (-)- EC, (-)-EGC, (-)- ECG y (-)-EGCG, se probaron las siguientes condiciones de análisis: longitud de onda (rango 190 a 900 nm); composición de fase móvil (metanol:agua, metanol:acetonitrilo, metanol:agua:ácido acético, metanol:agua:ácido fosfórico y acetonitrilo:agua (Her y Koel, 2003; Auger et al., 2004; Nishitani y Sagesaka, 2004) y temperatura (30, 35 y 40°C). Con lo determinado en las pruebas preliminares y, para establecer las mejores condiciones de análisis, se realizó un diseño experimental central compuesto 33. Las variables consideradas fueron: porcentaje de metanol (15, 18 y 20%), cantidad de ácido acético en agua (0, 0,1 y 0,3%) y temperatura (30, 35 y 40°C).
En el análisis de catequinas por CE se evaluaron diferentes condiciones, a saber: composición de la solución buffer, pH de esta solución, voltaje y temperatura. Se realizó un diseño experimental factorial 32 para evaluar el efecto que tuvieron sobre la resolución las concentraciones de los componentes de la solución buffer, ácido bórico (25, 35 y 45 mM), dihidrógeno fosfato de sodio (20 mM) y dodecil sulfato de sodio (60, 80 y 100 mM).
Discusión de resultados
Implementación de las metodologías de análisis de catequinas. Para la implementación de la metodología de análisis de catequinas por HPLC, se seleccionó la longitud de onda para el máximo de absorbancia de las catequinas a 210 nm.
Para seleccionar la composición de la fase móvil se realizaron pruebas variando los solventes de menor a mayor fuerza de elución, es decir, agua, metanol y acetonitrilo. Se evaluaron diferentes mezclas de solventes, a saber: metanol:agua, metanol:acetonitrilo, metanol:agua:ácido acético, metanol:agua:ácido fosfórico y acetonitrilo:agua (Figura 1).
Al emplear la fase móvil con mayor porcentaje de agua, se observó la separación de los cinco analitos, aunque el valor de resolución no fue completamente aceptable para los picos cromatográficos 1 y 2 (Figura 1). Con el propósito de reducir los tiempos de análisis, se evaluaron mezclas de solventes con menor retención, a saber: acetonitrilo: agua y acetonitrilo:metanol. Aunque se lograron tiempos de análisis más cortos, la resolución disminuyó drásticamente y se presentó la coelución de algunos picos.
La fase móvil con la que se obtuvo la resolución más alta, fue agua:metanol:ácido acético. Una vez establecida la composición de la fase móvil, se efectuó un diseño experimental central compuesto 33 para estudiar el efecto del porcentaje de metanol, cantidad del ácido acético y la temperatura del termostato.
El valor máximo de la variable de respuesta (resolución) en el diseño experimental central compuesto 33, se logró con 18% de metanol; 82% de agua:ácido acético (100:0.1) y la temperatura de 35 °C. En la Figura 2, se muestra un perfil cromatográfico típico de la mezcla patrón de (±)-C, (-)-EC, (-)-EGC, (-)-ECG y (-)-EGCG, obtenido bajo las condiciones descritas anteriormente.
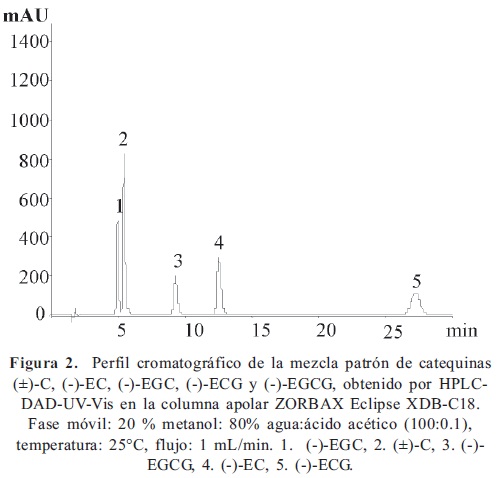
Para la implementación de la metodología de electroforesis capilar, se realizaron pruebas preliminares para determinar cuáles eran las variables que afectaban en mayor grado la separación de las cinco catequinas bajo estudio. De acuerdo con estos resultados, se determinó que los principales parámetros a evaluar en el diseño de experimentos eran los correspondientes a la composición de la fase móvil. Dado el carácter no iónico de las catequinas, se empleó como modalidad de electroforesis la llamada cromatografía electrocinética micelar (MEKC) (Kartsova y Ganzha, 2006); se utilizó como surfactante el dodecil sulfato de sodio (SDS), se probaron diferentes sustancias como componentes de la solución buffer, e.g. ácido bórico, tetraborato de sodio y dihidrógeno fosfato de sodio; se observó la separación parcial de las cinco catequinas (Figura 3). Teniendo en cuenta que la solución compuesta por la mezcla de NaH2PO4 y H3BO3 mostró los mejores resultados, se procedió a evaluar en un diseño de experimentos 32 el efecto de la concentración de ácido bórico y SDS en la solución buffer, manteniendo fija la concentración de dihidrógeno de sodio en la menor cantidad necesaria para lograr la separación de las 5 catequinas y evitar la posible precipitación de sales al interior del capilar por interacción de esta sustancia con metanol.
Como resultado del diseño experimental, en el que la variable de respuesta a evaluar fue la resolución entre los picos 1 y 2, se obtuvo que la solución buffer compuesta por dihidrógeno fosfato de sodio (20 mM), ácido bórico (35 mM) y SDS (80 mM) produjo la mejor separación. En la Figura 4 se presenta un electroforegrama de la mezcla de las cinco catequinas bajo estudio, obtenido empleando la metodología descrita. Los valores de parámetros como el voltaje aplicado al capilar (20 kV), el pH de la solución buffer (pH = 8,24) y la temperatura de análisis (20°C), se mantuvieron fijos.
Estudio comparativo entre las técnicas HPLC y CE. El estudio comparativo de las metodologías implementadas para la determinación de catequinas, se realizó teniendo en cuenta las figuras analíticas de mérito, el tiempo y el costo de análisis.
Las siguientes fueron las figuras analíticas de mérito determinadas: precisión (expresada como repetibilidad en tiempo de retención y área), porcentaje de recuperación, límite de detección, límite de cuantificación, linealidad (coeficiente de determinación) y sensibilidad (pendiente de la curva a un nivel de confianza del 95%). Los resultados se presentan en la Tabla 1.
Los límites de detección (LOD) y cuantificación (LOQ) fueron más bajos para HPLC excepto para la (-)-ECG. En general, para CE los LOD obtenidos, fueron más del doble de los encontrados para HPLC. Los mejores valores en cuanto a linealidad del método, evaluada en el rango de 0,5 a 64 mg/ L, se obtuvieron con HPLC. De manera similar, la sensibilidad asociada con la respuesta del detector, fue mayor para todas las catequinas en el DAD del equipo de HPLC empleado. Lo anterior indica que, en el análisis de catequinas bajo las condiciones establecidas, la mayor repetibilidad se alcanza con HPLC, hecho que coincide con reportes previos (Vanhoenacker et al., 2001; Nemenak et al., 2006).
El método de HPLC fue más reproducible en cuanto a tiempos de retención y áreas, de acuerdo con los CV % observados. Entonces, se puede afirmar que en el análisis de catequinas, las dos técnicas pueden emplearse para la cuantificación rutinaria de (±)-catequina, (-)- epicatequina, (-)-epigalocatequina, galato de (-)-epicatequina y galato de (-)-epigalocatequina, siguiendo las metodologías implementadas.
Aunque ambos métodos son eficientes para el análisis de las cinco catequinas, se observa que el tiempo de migración electroforético, para todas éstas, fue más corto (ca. 12 min), que el tiempo de retención (ca. 26 min), por HPLC. Sin embargo, para las primeras cuatro catequinas estudiadas por HPLC el tiempo fue menor que para CE (ca. 11 min). por tanto, en cuanto al tiempo de análisis, la selección entre las dos técnicas empleadas, estará establecida por cuáles analitos se requiere determinar.
El costo de análisis de las cinco catequinas en una muestra llega a ser mucho mayor por HPLC, debido a que se emplea un volumen hasta 60 veces mayor de solventes que en CE; además, el tiempo de análisis más largo representa costos más altos por uso del equipo y el tiempo del operador. Consecuentemente, HPLC presenta grandes desventajas desde los puntos de vista económico y ambiental, dados los altos costos de los solventes grado HPLC, tiempo de análisis y los problemas inherentes a la disposición de residuos químicos.
La metodología implementada por CE resultó ser bastante reproducible, repetible y sensible; sin embargo, no supera en estos tres aspectos a la que utiliza HPLC. No obstante, la técnica HPLC resultó ser menos ventajosa en cuanto a tiempos de análisis y costos. por lo tanto, se puede establecer que la decisión para escoger entre estas dos técnicas para el análisis de (±)-catequina, (-)-epicatequina, (-)- epigalocatequina, galato de (-)-epicatequina y (-)-galato de epigalocatequina, depende fundamentalmente de la disponibilidad de muestra, tiempo y reactivos, ya que ambos equipos, HPLC y CE, poseen precios similares.
Cuantificación de catequinas. Se analizaron los extractos de las cinco muestras de tres variedades de cacao (ICS- 39, TSH-565 e IMC-67) en cuanto a su contenido de catequinas, empleando las metodologías implementadas basadas en HPLC y EC. Los resultados se presentan en la Tabla 2. Las catequinas (-)-EGC, (-)-ECG y (-)-EGCG, no se hallaron en ninguno de los extractos por encima de los niveles mínimos de detección. Las catequinas (±)-C y (-)- EC se detectaron y cuantificaron en los cinco extractos. Mientras que las diferencias en la cuantificación de (±)-C por CE y por HPLC no fueron significativas, los resultados de la cuantificación de (-)-EC presentaron diferencias relativas entre 2,7 y 13,9%, siendo en todos los 5 casos el valor obtenido por CE inferior al resultado de HPLC. En la Figura 5 se observan un cromatograma y un electroforegrama representativo del extracto de la variedad TSH-565 de cacao estudiada.
Agradecimientos
Los autores agradecen a COLCIENCIAS por su apoyo financiero a través del centro de Excelencia CENIVAM (Contrato RC-432-2004) y al Laboratorio de Cromatografía de la Universidad Industrial de Santander.
Referencias
Auger, C.; Al-Awwadi, N.; Bornet, A.; Rouanet, J.; Gase, F.; Cros G.; Teissedre P. 2004. Catechins and procyanidins in mediterranean diets. Food Res Inter 37:233-245. [ Links ]
Bowser, M.; Frost, N.; Jing, M. 2010. Capillary electrophoresis. Anal Chem 82:4682-4698. [ Links ]
Castro, M.; López, J.; Gonzáles, M.; Barral, L. 2011. Development, validation and application of micellar capillary chromatography method for rutine analysis of catechins, quercetin and thymol in natural samples. Microchem J 99:461- 469. [ Links ]
Calderón, A.; Wright, B.; Hurts, J.; Breemen, R. 2009. screening antioxidants using LC-MS: Case study with cocoa. J Agric Food Chem 57:5693-5699. [ Links ]
Dalluge, J.; Nelson, B. 2000. Determination of tea catechins. J Cromatogr A 880:411-424. [ Links ]
Folch-Cano, C.; Jullian, C.; Speisky, H.; Olea-Azar, C. 2010. Antioxidant activity of inclusion complexes of tea catechins with â-cyclodextrins by ORAC assays. Food Res Int 43:2039- 2044. [ Links ]
Her, M.; Koel, M. 2003. Separation of polyphenolic compounds extracted from plant matrices using capillary electrophoresis. J Chromatogr A 990:225-230. [ Links ]
Kartsova L.; Ganzha O. 2006. Electrophoretic separation of tea flavanoids in the modes of capillary zone electrophoresis and micellar electrokinetic chromatography, Russ J Appl Chem 79:1110-1114. [ Links ]
Niemenak, N.; Rohsius, C.; Elwers, S.; Ndoumou D. O.; Lieberei R. 2006. Comparative study of different cocoa (Theobroma cacao L.) clones interms of their phenolics and anthocyanins contents. J Food Composition Anal 19:612- 619. [ Links ]
Nishitani, E.; Sagesaka Y. 2004. Simultaneous determination of catechins, caffeine and other phenolics compounds in tea using new HPLC method. J Food Comp Anal 17:675-685. [ Links ]
Putte, L. B.; Hollman, P. 2000. Catechin contents of foods commonly consumed in the Netherlands. 2. Tea, Wine, Fruit Juices, and Chocolate Milk. J Agric Food Chem 48:1752- 1757. [ Links ]
Rice-Evans, C. 1996. Structure-antioxidant activity relationships of flavonoids and phenolic acids. Free Radical Biol Med 20(7):933-956. [ Links ]
Tsao, R.; Deng, Z. 2004. Separation procedures for naturally occurring antioxidant phytochemicals. J Chromatogr B 812:85-99. [ Links ]
Valls, J.; Millán, S.; Martí, M.; Borràs, E.; Arola, L. 2009. Advanced separation methods of food anthocyanins, isoflavones, and flavonols. J Chromatogr A 1216(43):7143- 7172. [ Links ]
Vanhoenacker, G.; Villiers, A.; Lazou, K.; De Keukeleire D.; Sandra P. 2001. Comparison of high-performance liquid chromatographymass spectroscopy and capillary electrophoresis-mass spectroscopy for the analysis of phenolic compounds in diethyl ether extracts of red wines. J Chromatogr A 54:309-315. [ Links ]
Yilmaz, Y. 2006. Novel uses of catechins in foods. Trends in Food Sci & Tech 17:64-71. [ Links ]
Recibido: agosto 5 de 2011. Aceptado para su publicación: agosto 30 de 2011.
