










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista de la Academia Colombiana de Ciencias Exactas, Físicas y Naturales
versão impressa ISSN 0370-3908
Rev. acad. colomb. cienc. exact. fis. nat. vol.35 no.136 Bogotá jul./set. 2011
QUÍMICA DE MATERIALES
Oliva Primera-Pedrozo, Fredy Colpas-Castillo1, Edgardo Meza-Fuentes2 Roberto Fernández-Maestre3
1 Grupo de carboquímica.
2 Grupo de estudios en materiales y combustibles.
3 Grupo de Medio Ambiente y Química Aplicada.
1 Autor correspondiente: rfernandezm@unicartagena.edu.co. Campus de Zaragocilla, Programa de Química, Cartagena, Colombia.
Resumen
Se prepararon carbones activados con bagazo de caña de azúcar (BC) y zuro de maíz (ZM) para adsorber cadmio y plomo. BC y ZM fueron carbonizados a 400°C por 1 hora, activados con ácido fosfórico y modificados con ácido nítrico o peróxido de hidrógeno y posterior calentamiento. La activación aumentó el área superficial de los carbones (5 m2/g a 778 m2/g para el ZM y 3 m2/g a 369 m2/g para BC). Los grupos oxigenados se analizaron por IR-DRIFTS. La acidez de los carbones estuvo entre 1.36 y 2.12 meq/g, el pH entre 2.9 y 6.5, y las capacidades de intercambio iónico hasta 0.070 meq/g (absorción atómica).
Palabras clave: bagazo de caña, zuro de maíz, cadmio, plomo, carbón activado, carbonizado.
Abstract
Activated carbons were prepared with sugar cane bagasse (BC) and corn cob (ZM) to adsorb aqueous cadmium and lead. BC and ZM were carbonized at 400°C for 1 hour, activated with phosphoric acid and modified with nitric acid or hydrogen peroxide and subsequent heating. Activation increased the surface area of carbons (5 m2/g to 778 m2/g for ZM and 3 m2/g to 369 m2/g for BC). Oxygenated groups were analyzed by IR-DRIFTS. The acidity of carbons was between 1.36 and 2.12 meq/g, pH between 2.9 and 6.5, and ion exchange capacities up to 0.070 meq/g (atomic absorption).
Key words: sugar cane bagasse, corn cob, cadmium, lead, activated carbons, carbonized.
Introducción
El carbón en sus diferentes formas juega un papel importante en muchas industrias. La mayoría de carbones industriales son producidos a partir de materiales carbonosos naturales como huesos de frutas, lignitos, carbón de hulla, turba, etc., a los cuales se les aplica un tratamiento térmico en ausencia de aire a temperaturas inferiores a 900ºC. El nombre “Carbones Activados” es usado para denominar los materiales carbonosos que han sido tratados con un agente químico o físico activante; la activación química aumenta la porosidad y forma diferentes grupos funcionales sobre la superficie del carbón (Marsh et al. 1997).
Activación química. La materia carbonácea se impregna con una disolución concentrada de agentes químicos y se carboniza entre 350 y 900ºC en atmósfera inerte (Rodríguez-Reinoso & Molina-Sabio, et al. 1998). Los agentes de activación química más utilizados son el cloruro de zinc, el ácido fosfórico y el hidróxido de potasio. El cloruro de zinc ha sido desplazado por el ácido fosfórico por problemas de corrosión, bajos rendimientos y presencia de zinc residual en el producto final. Los carbones activados obtenidos con estos agentes son generalmente mesoporosos, de baja densidad y poca resistencia a la abrasión. Estas características los hacen aplicables para fase líquida pero no para fase gaseosa. Otros materiales para activar carbones son: hidróxido de sodio (Hao & Wang, 2010), sulfato, hipoclorito y fosfato de sodio, fosfato y cloruro de calcio (Rajakovic & Ristic, 1996) e hidróxido de calcio, cloruro y sulfato manganoso, ácidos bórico, nítrico (ShamsiJazeyi & Kaghazchi, 2010), sulfúrico y fosfórico (Rosas et al. 2009), cloruros férrico (Cui & Turn, 2009) y de zinc (Li et al. 2010), sulfuro, tiocianato y carbonato de potasio, azufre, dióxido de manganeso, dolomita, cianuros, peróxido de hidrógeno (Gan & Wu, 2008), etanotiol (Kim et al. 2011), 8-Hidroxiquinolina (Garoma et al. 2010), ácidos cítrico y tartárico, cloruro de bario (Rajakovic & Ristic, 1996), y gases como ozono (He et al. 2008), dióxido de carbono (Singh & Lal, 2010), dióxido de azufre y sulfuro de hidrógeno (Macias-García et al. 2003), cloro (Kim et al. 2011), etc.
Materia prima. Los carbones industriales son producidos en su mayoría a partir de materiales carbonosos naturales como carbón y material de desecho vegetal como madera, semillas y podas. Los materiales de partida para obtener carbón activado también pueden ser: algas, cáscara de arroz, lignina, aserrín, cáscara de semilla de algodón, lignito, lodos ácidos de petróleo, madera, carbón mineral, huesos de frutas, melaza, carbón vegetal, desperdicios de pasta de papel, negro de humo, residuos de carne y sangre, olotes, huesos, desechos de destilería, pescado, cereales, cuesco de palma africana, turba, carbohidratos, coque de petróleo, granos de café, hollín, cáscara de nueces, grafito (Arpa et al. 2000; Burns et al. 1999), cáscara de coco (Song et al. 2010), cáscara de frutas (Tongpoothorn et al. 2011), palma de aceite (Navarrete et al. 2005), y residuo de tabaco (Kilic et al. 2011). Estas materias primas deben tener alto contenido de carbono y bajo contenido de material mineral, y ser de fácil activación, alto rendimiento, baja degradación durante su almacenamiento y bajo costo.
Estructura de los carbones activados. Los carbones activados tienen una estructura porosa con pequeñas cantidades de heteroátomos, principalmente oxígeno y pueden contener hasta 20% en peso de materia mineral, expresado como contenido de cenizas; la naturaleza de las cenizas depende del material de partida del carbón activado; normalmente un carbón activado está compuesto de placas de 2-5 nm de altura constituidas por unidades de 5 a 15 anillos aromáticos condensados, semejantes a la estructura de un grafito (Marsh et al. 1997; López-Ramón et al.1999). El carbón activado adsorbe una gran cantidad de compuestos orgánicos e inorgánicos de fases líquidas o gaseosas. Estos poros pueden dividirse en macroporos (diámetro mayor de 500 Å), mesoporos (entre 500 y 20 Å) y microporos (menor de 20 Å) (Juntgen, 1986; Castilla et al. 2000). La distribución del tamaño del poro se obtiene utilizando isotermas de adsorción, principalmente por el método BET (Haenel, 1982). El área superficial, la distribución, volumen y tamaño de poro, la superficie química, la distribución y densidad de la partícula, la resistencia al calor, la dureza, el contenido de cenizas y otros parámetros que caracterizan al carbón activado dependen de la materia prima y de las condiciones de carbonización y activación (Marsh et al. 1997). La superficie química juega un papel importante ya que los heteroátomos y los grupos funcionales oxigenados de la superficie pueden actuar como sitios activos.
La química de superficie de los materiales carbonosos esta básicamente determinada por el carácter básico o ácido de su superficie. Los tratamientos con agentes oxidantes fijan en su superficie complejos oxigenados tales como carboxilos, lactonas, fenoles, cetonas, quinonas, alcoholes y éteres, adecuados para la complejación de metales (López-Ramón et al. 1999; Castilla et al. 2000). Los carbones activados han sido aplicados a la adsorción de contaminantes como gases e iones metálicos de plomo (Li et al. 2010; Song et al. 2010), cadmio (Kim et al. 2011; Garoma et al. 2010), y otros metales pesados.
En este trabajo se prepararon carbones activados para adsorber cadmio y plomo con bagazo de caña de azúcar (BC) y zuro de maíz (ZM) mediante carbonización a 400°C, activación con ácido fosfórico y oxidación con ácido nítrico o peróxido de hidrógeno.
Experimental
Los carbones preparados fueron codificados como ZM (provenientes del zuro de maíz) y BC (provenientes del bagazo de caña).
Recolección y preparación de los materiales vegetales de partida. El bagazo de caña de azúcar se recogió en el mercado de Bazurto, Cartagena, y el zuro de maíz en el corregimiento de Palo Alto (Sucre), en Colombia. El bagazo y el zuro fueron secados a 105ºC por 24 horas y luego fueron triturados.
Carbonización. Quinientos gramos de los materiales vegetales se calentaron (usando el montaje de la Figura 1) en fracciones de 100 g desde 30ºC hasta 400ºC (10ºC/min) en atmósfera de nitrógeno (110 mL /min) y fueron mantenidos a estas condiciones por 60 minutos (Hsisheng & Hsu, 1998). El carbón obtenido se molió y se tamizó a un tamaño de partícula de 125 a 300 micrómetros. Estos carbones fueron codificados como ZM-C (provenientes del zuro de maíz) y BC-C (provenientes del bagazo de caña).
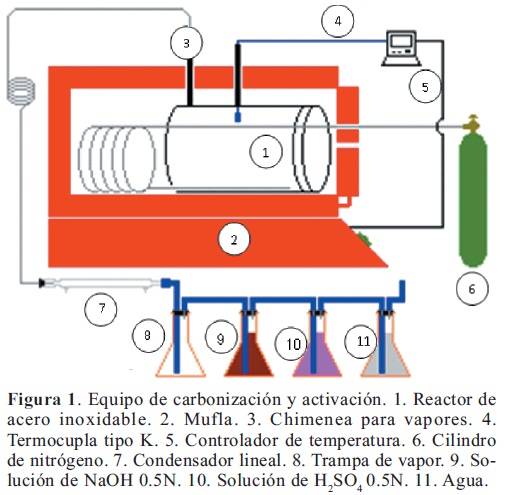
Activación química de los carbones con ácido fosfórico. Veinte gramos de cada material carbonizado se sometieron al siguiente tratamiento (Hsisheng & Hsu, 1998; Huidobro et al. 2000): Se impregnaron con ácido ortofosfórico al 21% p/v (agente activante) con agitación constante por 4.5 horas, se secaron a 105°C por 24 horas, se calentaron en atmósfera de nitrógeno (flujo de 110 mL / min) desde 30ºC hasta 450 ± 5ºC (10ºC/min) y se mantuvieron a estas condiciones por 120 minutos. Estos carbones fueron codificados como ZM-A y BC-A. Estos carbones activados fueron modificados mediante tratamiento térmico a 600°C, y oxidaciones con ácido nítrico y peróxido de hidrógeno tal como se describe a continuación.
Tratamiento térmico de los carbones activados. Veinte gramos de los carbones activados ZM-A o BC-A se calentaron desde 30ºC hasta 600ºC (10ºC/min) en atmósfera de nitrógeno (110 mL /min) y fueron mantenidas a estas condiciones por 60 minutos. Estos carbones activados fueron codificados como ZMΔ y BCΔ.
Oxidación con ácido nítrico de los carbones activados. Veinte gramos de los carbones activados ZM-A o BCA se sometieron al siguiente tratamiento: Se agregaron 250 ml de HNO3 al 15% y se digirieron por 30 minutos, se secaron a 105 ºC por 24 horas, se calentaron en atmósfera de nitrógeno (flujo de 110 mL /min) desde 30ºC hasta 600 ± 5ºC (10ºC/min) y se mantuvieron a estas condiciones por 60 minutos. Estos carbones activados fueron codificados como ZM-HNO3 y BC-HNO3.
Oxidación con peróxido de hidrógeno de los carbones activados. 20 g de los carbones activados ZM-A o BCA se sometieron al siguiente tratamiento: se agregaron 250 ml de H2O2 al 10% y se digirieron por 30 minutos, se secaron a 105ºC por 24 horas, se calentaron en atmósfera de nitrógeno (flujo de 110 mL /min) desde 30ºC hasta 600 ± 5ºC (10ºC/min) y se mantuvieron a estas condiciones por 60 minutos. Estos carbones activados fueron codificadas como ZM-H2O2 y BC-H2O2.
Los materiales vegetales sometidos a carbonización, activación u oxidación se retiraron de la mufla cuando la temperatura era menor de 50°C, se lavaron alternativamente con agua caliente y fría hasta que la conductividad de las aguas de lavado fue muy similar a la conductividad del agua destilada usada para lavar, y se secaron a 105ºC por 12 horas (Huidobro et al. 2000).
Análisis Próximo. La humedad se determinó por la pérdida de peso al calentar los carbones a 150°C por 3 horas en una estufa Precision Scientific Modelo 18 siguiendo la norma ASTM – D 3173-87. El porcentaje de materia volátil se determinó por la pérdida de peso cuando los carbones secados a 150°C fueron calentadas a 900 ± 5°C por 7 minutos en atmósfera de nitrógeno en un horno Ohio Thermal siguiendo la norma ASTM – D 3175-99. El contenido de cenizas se calculó con el residuo después de quemar los carbones (previamente secados a 150°C por 3 horas) a 650 ± 5°C en atmósfera de aire hasta peso constante en un horno Ohio Thermal siguiendo la norma ASTM – D 3174-97. El carbono fijo es aquel que queda después de calentar a 650°C en atmósfera de N2 y se calculó por diferencia, Carbono fijo (%) = 100 – (% materia volátil + % cenizas).
Análisis último. El análisis último comprendió la determinación de carbono, hidrógeno azufre y nitrógeno, y el cálculo del oxígeno por diferencia y se realizó en un horno Lindberg siguiendo la norma ASTM – D 3178-89. La determinación de carbono e hidrógeno se realizó por pirólisis de los carbones en un sistema cerrado determinando los productos de la combustión, agua y dióxido de carbono, después de la oxidación completa. En este método, el carbono y el hidrógeno se determinaron por el incremento de peso de las trampas de CO2 y agua (Figura 1).
El nitrógeno se determinó en los laboratorios de Ingeominas, Cartagena. El nitrógeno de los carbones se convirtió en sales de amonio por digestión con ácido sulfúrico concentrado en una mezcla catalizada con sulfato de potasio a alta temperatura. Estas sales se disolvieron en una solución alcalina caliente de la cual el amonio se removió por destilación y se determinó por titulación ácido- base recogiéndolo en un recipiente con una cantidad medida de ácido clorhídrico. En la determinación de azufre total de los carbones se utilizó el método de la bomba lavadora (SCFI, 1982). En este método, los carbones se calcinan a 800°C por 90 minutos con una mezcla de óxido de magnesio y carbonato de sodio que precipita el azufre como BaSO4 a partir del cual se calcula el contenido de azufre. El porcentaje de oxígeno se calculó por diferencia.
Análisis de grupos funcionales oxigenados por espectroscopia infrarroja de reflectancia difusa con transformada de Fourier (DRIFTS). Los espectros DRIFTS fueron tomados de los carbones previamente secos (105°C por 12 horas) en un espectrómetro Nicolet 510P FT-IR, promediando 150 barridos con una resolución de 4 cm-1 en el rango de 800 cm-1 a 4000 cm-1. Los carbones fueron pulverizados y tamizados utilizando mallas de 200. Estos espectros fueron tomados en el Laboratorio de Carbones de la Universidad Nacional de Colombia.
pH de los carbones. Se mezclaron 1.000 g de cada carbón con 20.00 mL de agua destilada libre de CO2 (Castilla et al. 2000) en recipientes de vidrio y se agitaron a temperatura ambiente por 24 horas; luego, se midió el pH de las suspensiones acuosas con un electrodo de vidrio (Orion 940).
Caracterización textural de los carbones. Se midieron la superficie especifica (área total, área de microporos y área externa) utilizando la ecuación de BET, y la porosidad evaluada con el volumen de microporos (utilizando curvas t). Esta información se consiguió de las isotermas de adsorción de nitrógeno obtenidas en las siguientes condiciones: Desgasificado del carbón durante toda la noche a 300 ºC, peso de carbón 0.1000-0.2000 g, adsorbato nitrógeno, temperatura de adsorción 77K, rango de presión relativa 0.05-0.15 (para carbones activados), tiempo de equilibrio 25 s, velocidad de evacuación 300 mm Hg/min y tiempo de evacuación 1 min (Gregg & Sing, 1991). Estudios previos muestran que los carbones activados son esencialmente microporosos de acuerdo con la forma de las isotermas y que el modelo BET no cumple adecuadamente en el intervalo 0.05-0.35 de p/po (Gregg & Sing, 1991). por tal razón se escoge una región al principio de la isoterma donde el modelo BET arroje correlaciones de 0.999 o mejores, que es equivalente al rango de presiones relativas de 0.05-0.15 p/po.
Medida del área superficial. A cada una de los carbones activados se les determinó el área superficial específica por el método BET utilizando un equipo Micromeritics Gemini 2375 usando como adsorbato nitrógeno a 77 K, entre 0.01 y 0.1 p/po.
Determinación de grupos oxigenados básicos y ácidos por el método de Boehm (Boehm, 1994). Para los grupos oxigenados ácidos, se pesaron ~0.5000 g de cada carbón y se agitaron a temperatura ambiente por 24 horas con 50.00 mL de una solución de NaOH 0.1000 N. Luego se filtró el carbón y la solución se tituló potenciométricamente con HCl 0.1000 N para determinar la cantidad de reactivo consumido. Para los grupos oxigenados básicos se repitió el procedimiento anterior pero se usó ácido clorhídrico 0.1000 N para la agitación y NaOH 0.1000 N para la titulación.
Adsorción de plomo y cadmio. En cada experimento se pesaron aproximadamente 0.2000 gramos de cada carbón y se les adicionaron 50.00 mL de las soluciones de Cd(II) y Pb(II) de 18.0 ppm y se agitaron a 120 rpm por 18 horas a temperatura ambiente. Luego se filtró la solución y los metales se determinaron por espectroscopia de absorción atómica con llama de acetileno-aire. Se prepararon curvas de calibración de patrones de 0.050, 0.100, 1.000, 2.000 y 5.000 ppm para Pb (283.3 nm) y de 0.100, 0.200, 0.500, 1.000 y 2.500 ppm para Cd (228.8 nm) con un coeficiente de correlación mínimo de 0.999; los patrones y las soluciones de intercambio se prepararon con estándares certificados de 1000.0 ppm (Sigma) de cada uno de los metales.
Análisis estadístico. Todos los cálculos estadísticos se realizaron con la ayuda de statgraphics plus versión 3.0. Se realizaron pruebas anova encontrar diferencias significativas entre las áreas superficiales y las capacidades de adsorción de plomo y cadmio de todos los carbones.
Resultados y discusión
Se obtuvieron carbonizados y carbones activados a partir del zuro de maíz y bagazo de caña de azúcar con altas capacidades de adsorción de plomo y cadmio; algunos carbones removieron más del 99% de los metales en soluciones con una concentración de 15 ppm.
Carbonización del zuro de maíz y bagazo de caña de azúcar. Durante este proceso las unidades de celulosa, principal componente de los materiales vegetales de partida sufren depolimerización transformándose en unidades más pequeñas, se desprende materia volátil y se produce un sólido más rico en carbono (Rodríguez-Reinoso & Molina-Sabio, 1998). La Tabla 1 muestra los análisis próximo y último de los materiales vegetales de partida, carbonizados y carbones activados. El contenido de materia volátil y de carbono fijo para ambos materiales de partida es similar, arrojando rendimientos similares en la producción de carbones activados. El contenido de oxígeno y carbono en los materiales vegetales de partida también es muy similar lo que hace que los carbones producidos tengan propiedades parecidas. El proceso de carbonización aumentó significativamente el contenido de carbono de 47 a 74% y de 49 a 80% para BC y ZM respectivamente, disminuyó los porcentajes de nitrógeno, oxígeno, hidrógeno y materia volátil, y aumentó el carbono fijo y las cenizas para ambos carbones (Tabla 1).
Activación de los carbonizados. En los carbones activados, el contenido de carbono aumentó con una disminución proporcional de otros constituyentes químicos (Tabla 1). Este aumento del contenido de carbono se debe al carácter deshidratante del ácido fosfórico, que facilitó la pérdida de hidrógeno y oxígeno, y convirtió unidades carbonadas grandes en unidades más pequeñas con mayor riqueza en carbono (Rodríguez-Reinoso & Molina-Sabio, 1998). El ácido fosfórico aumenta la aromaticidad del carbón, la pérdida del carácter alifático y la formación de un sólido rígido entrecruzado (Hsisheng & Hsu, 1998; Huidobro et al. 2000). El bagazo de caña de azúcar y el zuro de maíz están compuestos básicamente por lignina, celulosa y hemicelulosa; con la carbonización a 400°C solo la celulosa y hemicelulosa se depolimerizaron en unidades más pequeñas para dar origen a una gran masa de carbono fijo ya que la lignina se descompone a temperaturas mayores de 200-500°C (Berkowitz, 1957; Kudo & Yoshida, 1957). Cuando se impregnó el carbonizado con ácido fosfórico se facilitó la hidrólisis de la lignina, que aún estaba presente en el carbonizado, de forma que aumentó el rendimiento del proceso y a la vez se desarrolló porosidad (Rodríguez- Reinoso & Molina-Sabio, 1998).
Después de la pérdida de agua, el ácido fosfórico se compone de una mezcla de ácidos polifosfóricos, incluyendo especies como H3PO4 , H4P2O7 y H5P3O10 y otras especies, de fórmula general Hn+2PnOn+1 (Hsisheng & Hsu, 1998). A la temperatura de activación, los ácidos polifosfóricos no sufren descomposición o evaporación ya que esta ocurre entre 600-800°C (Hsisheng & Hsu, 1998; Huidobro et al. 2000) por lo que en la activación no ocurrió una notable pérdida de peso como en la carbonización. Después de la activación de los materiales carbonizados, hubo un aumento en su contenido de cenizas; este aumento puede ser atribuido a las interacciones entre el fósforo del ácido fosfórico y la materia mineral proveniente del bagazo de caña de azúcar y zuro de maíz (Hsisheng & Hsu, 1998).
Modificaciones de los carbones activados. Las pérdidas de peso para cada uno de los tratamientos a los cuales fueron sometidos los carbones fueron ZM-C, 62%; ZM-A, 21%; ZM–H2O2, 22%; ZM–HNO3, 54%; ZMΔ, 10%; BC-C, 65%; BC-A, 23%; BC–H2O2, 22%; BC–HNO3, 57%; y BCΔ, 11%. La carbonización causó una pérdida significativa de peso, mayor del 62% del peso del bagazo y del zuro; de igual forma, el tratamiento de los carbones activados con ácido nítrico causó una pérdida de peso mayor del 50%. La pérdida de peso originada por el tratamiento térmico de los carbones activados fue menor que la ocasionada por las oxidaciones con ácido nítrico y peróxido de hidrógeno; lo anterior indica que los agentes oxidantes en el proceso de oxidación debilitaron las paredes de los poros generando pérdida de carbón en la matriz carbonosa.
Análisis infrarrojo. La Interpretación de los espectros infrarrojos con transformada de Fourier con reflectancia difusa (DRIFTS) es complicada por el hecho que cada grupo origina varias bandas por lo que cada banda podría incluir contribuciones de varios grupos (Figueiredo et al. 1999). La Figura 2 muestra el espectro DRIFTS, números de onda (ν, cm-1) vs. porcentaje de transmitancia, de BC-C. se encontraron bandas a 1200 cm-1 (C-O en éteres); 1049-1276 cm-1 (alcoholes); 1100 cm-1 y 3400 cm-1 (C-OH fenólico); 1585-1600 cm-1 (C=C aromático); 2900 cm-1 (C-H alifático); y 1150 cm-1, 1700 cm-1, 3400 cm-1 (ácidos carboxílicos); en las regiones ~3400 y ~1700 cm-1 los carbones oxidados presentaron bandas más pronunciadas que los originales indicando el aumento de grupos oxigenados a causa de la oxidación. La banda alrededor de 1590 cm-1, que producen todos los carbones, puede ser atribuida a estiramientos de dobles enlaces de anillos aromáticos conjugados a grupos carbonílicos.
Acidez de los carbones. La Tabla 2 muestra el contenido de grupos ácidos y básicos de los carbones preparados; el pH de las suspensiones acuosas de los carbones fue ácido, (2.9 a 6.5), lo cual significa que estos carbones mostraban una predominancia de grupos funcionales ácidos en su superficie, importantes para el proceso de adsorción de metales. El post-tratamiento térmico a 600 °C no modificó la acidez de los carbones pues los grupos básicos se forman a temperaturas mayores de 800°C (Studebaker, 1957); esto sugiere que los sitios básicos de superficie son esencialmente del tipo de Lewis, asociado con regiones π ricas en electrones (Lopez-Ramon et al. 1999); en general, las oxidaciones incrementaron la acidez de ZM-A y BC-A, especialmente la oxidación con peróxido de hidrógeno; en ZMΔ y BCΔ se mantuvo la acidez total presente en ZM-A y BC-A. La Tabla 2 también nos muestra que: a. El carbón con mayor área superficial fue ZM-A (área total 778.3 ± 0.7 m2/g); b. Los carbones activados originarios de zuro de maíz presentaron mejores parámetros texturales que los correspondientes a bagazo de caña; y c. La basicidad total (meq HCl/g) fue cero para todos los carbones indicando que los carbones eran ácidos.
Análisis de las isotermas. Las isotermas de adsorción de nitrógeno de los materiales carbonizados (no mostradas) no se ajustaron a los tipos de isotermas de BET y mostraron que no existió adsorción de nitrógeno. En cambio, todas las isotermas de los carbones activados fueron del tipo I (BET) (Gregg & Sing, 1991). En casi todos los carbones activados y modificados, 70% del volumen de poros fue llenado a presiones relativas de 0.1 (p/po) (Figura 3) indicando que estas muestras eran altamente microporosas (diámetro de poros menor de 20 A) (Juntgen, 1986); de acuerdo a las isotermas, los carbones con mayor área superficial fueron ZM-A y BC-H2O2.
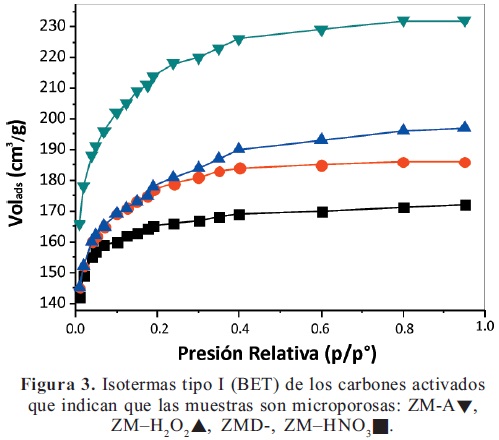
Influencia de las modificaciones en el área superficial. La activación con ácido fosfórico desarrolló la porosidad de los carbones y aumentó el área superficial (Tabla 2); esta porosidad puede atribuirse al carácter deshidratante del ácido fosfórico que promovió reacciones de hidrólisis ácida (Marsh et al. 1997). Las modificaciones alteraron las áreas superficiales específicas de los carbones activados de una forma diferente lo que indica diferencias importantes de las estructuras y características de los materiales vegetales de partida.
El material vegetal que mejor desarrolló sus propiedades adsortivas durante la carbonización y la activación fue el zuro de maíz con una área total de 778 m2/g (Tabla 2) comparable a la de carbones activados comerciales (Norit, 1999).
Adsorción de cadmio y plomo en los carbones. Puesto que la naturaleza química de los carbones es similar, las diferencias encontradas en la capacidad de adsorción podrían ser debidas a características superficiales como área y volumen. Los resultados de adsorción de plomo y cadmio están dados en la Tabla 3 y la Figura 4.
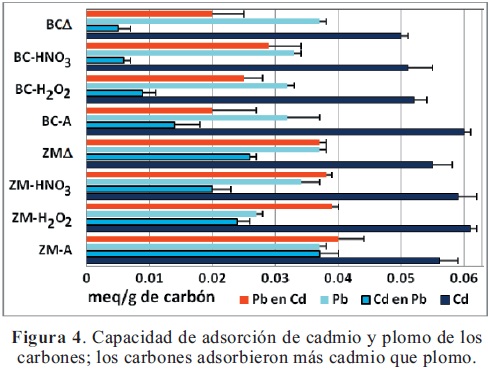
Adsorción de plomo. Las mayores capacidades de adsorción de plomo la mostraron ZM-A, ZMΔ y BCΔ (~0.037 meq/g) y la menor la mostró ZM-H2O2 (0.027 meq/ g). Los capacidades de adsorción de plomo de los demás carbones (~0.033 meq/g) fueron estadísticamente iguales; la ubicación de grupos oxigenados a la entrada de los poros, que impiden la difusión de los metales al interior de los mismos, se ha usado para explicar la menor adsorción de metales por parte de algunos carbones (Pradhan &Sandle, 1999); esto podría indicar porque algunos carbones con mayor acidez y área de microporos tienen una baja capacidad de adsorción.
Adsorción de cadmio. La mayor capacidad de adsorción de cadmio la mostró ZM-H2O2 (0.061 meq/g), pero fue estadísticamente igual a las de ZM-A, ZM-HNO3, y BC-A (~0.059 meq/g); la menor capacidad de adsorción de cadmio la mostraron BC-H2O2, BC-HNO3 y BCΔ (~0.051 meq/g) (Tabla 3).
En general, la capacidad de adsorción de Cd fue mayor cuando estaba solo que en presencia de plomo, (Figura 4). Además, los metales fueron mejor removidos cuando estaban solos, debido a que no tenían que competir con el otro metal por los sitios activos de la superficie del carbón; la Figura 4 muestra que todos los carbones presentaron mayor selectividad para adsorber cadmio que plomo (p < 0.05); esto se atribuye a que el ion cadmio es más pequeño que el plomo y por lo tanto va a tener mayor velocidad de difusión, mayor acceso a los poros y mayor posibilidad de ser adsorbido. La mezcla de los metales afectó más la adsorción de Cd que la de Pb (Figura 4); la mayor adsorción de Cd y Cd en presencia de Pb la mostró ZM-H2O2 y la mayor adsorción de Pb y Pb en presencia de Cd la presentaron ZM-A y ZM-H2O2. La capacidad de adsorción de metales dependió de una forma compleja de una combinación de varias propiedades de los carbones.
Conclusiones
Se prepararon carbones activados con bagazo de caña de azúcar y zuro de maíz para adsorber cadmio y plomo de soluciones acuosas. El bagazo de caña y zuro de maíz fueron carbonizados, activados con ácido fosfórico y oxidados con ácido nítrico o peróxido de hidrógeno y posteriormente calcinados en atmósfera de nitrógeno. La activación con ácido fosfórico causó un aumento en el área superficial. En general, una mayor cantidad de grupos oxigenados fue fijada por el oxidante peróxido de hidrógeno y se observó un aumento en la cantidad de grupos ácidos con las oxidaciones. Se obtuvieron carbones activados con altas capacidades de adsorción de cadmio y plomo que por lo general dependieron de la porosidad de los carbones. Los carbones activados fueron más selectivos para adsorber cadmio que plomo. En general, los metales se removieron con mayor facilidad cuando se encontraban solos que combinados y la adsorción de plomo fue afectada más por la presencia de cadmio que lo contrario. La implementación de este método de activación en dos etapas no favoreció la capacidad de adsorción de los carbones activados.
Referencias
Arpa, C., E., Basyilmaz, S., Bektas, O., Genc, Y., Yurum. 2000. Cation exchange properties of low Turkish coals: removal of Hg, Cd and Pb from wastewater. Fuel Process. Technol 68(2):111-120. [ Links ]
Berkowitz, N. 1957. On the Differential Thermal Analysis of Coal, Fuel 36:355-373. [ Links ]
Boehm, H. P. 1994. Some aspects of the surface chemistry of carbon blacks and other carbons. Carbon 32(5):759-769. [ Links ]
Burns, C. A., P. J., Cass, I., Harding, H., Russell, J., Crawford. 1999. Adsorption of aqueous heavy metals onto carbonaceous substrates. Colloids and surfaces 155(1):63-68. [ Links ]
Castilla, M., M. V., Lopez-Ramon, F., Carrasco-Marin. 2000. Changes in surface chemistry of activated carbons by wet oxidation. Carbon 38(14):1995-2001. [ Links ]
Cui, H., S. Q., Turn. 2009. Adsorption/desorption of dimethylsulfide on activated carbon modified with iron chloride. Appl Catal B Environ 88(1-2):25-31. [ Links ]
Figueiredo, J. L., M. F. R., Pereira, M. M. A., Freitas, y J. J. M., Orfao. 1999. Modification of the surface chemistry of activated carbons. Carbon 37(9):1379-1389. [ Links ]
Gan, T., K., Wu. 2008. Sorption of Pb (II) using hydrogen peroxide functionalized activated carbon, Colloids Surf. A: Physicochem Eng Aspects 330(2-3):91-95. [ Links ]
Garoma, T., S., Shah, B., Badriyha. 2010. Removal of Cadmium, Copper, and Zinc from Aqueous Solution by Activated Carbon Impregnated with 8-Hydroxyquinoline. J Environ Sci Eng 4(5):14-21. [ Links ]
Gregg, S. J., K. S. W., Sing. 1991. Adsorption, surface area and porosity. Academic press Inc. New York. [ Links ]
Haenel, M. W. 1982. Chemistry of Coal Utilization, Angew Chem 94(6):469-470. [ Links ]
Hao, Y. N., X. M., Wang. 2010. Adsorption of Basic Fuchsin onto Xanthoceras Sorbifolia Bunge Crust Activated Carbon Developed by Sodium Hydroxide. Adv Mater Res 156-157:272-278. [ Links ]
He, X-C., J-T., Zheng, W-Z., Yu, G-C., Wang X-F., Qu. 2008. Modification of activated carbon by ozone and its thermodynamics and kinetics of the adsorption for thiophene. Acta Petrolei Sinica (Petrol. Proc. Section). 2008-04. [ Links ]
Hsisheng, T., T-S., Yeh, L-Y., Hsu. 1998. Preparation of Activated Carbon from Bituminous Coal with Phosphoric Acid Activation. Carbon 36(9):1387-1395. [ Links ]
Huidobro, A., A. C., Pastor, F., Rodríguez-Reinoso. 2000. Preparation of Activated Carbon Cloth from Viscous Rayon. Part IV. Chemical Activation. Carbon 39(3):389-398. [ Links ]
Juntgen, H. 1986. Activated Carbon as Catalyst Support. Fuel 65(10):1436-1446. [ Links ]
Kilic, M., E., Apaydin-Varol, A. E., Pütün. 2011. Adsorptive removal of phenol from aqueous solutions on activated carbon prepared from tobacco residues: Equilibrium, kinetics and thermodynamics. J Hazard Mat 189(1-2):397-403. [ Links ]
Kim, D., Y. W., Jung, S., Kwon, J-W., Park. 2011. Adsorption of cadmium(II) from aqueous solutions by thiol-functionalized activated carbon. Water Sci. Technol.: Water Suppl. 11(1):61-66. [ Links ]
Kudo, K., E., Yoshida. 1957. On the decomposition process of wood constituents in the course of carbonization. J. Jap. Wood Res Soc 3(4):125-127. [ Links ]
Li, Y., Q., Dua, X., Wang, P., Zhang, D., Wang, Z., Wang, Y., Xia. 2010. Removal of lead from aqueous solution by activated carbon prepared from Enteromorpha prolifera by zinc chloride activation. J Hazard Mat 183(1-3):583-589. [ Links ]
Lopez-Ramon, M. V., F., Stoeckli, C., Moreno-Castilla, F., Carrasco-Marin. 1999. On the characterization acidic and basic surface sites on carbons by various techniques. Carbon 37(8):1215-1221. [ Links ]
Macias-Garcia, A., V., Gomez-Serrano, M. F., Alexandre-Franco, C., Valenzuela-Calahorro. 2003. Adsorption of cadmium by sulphur dioxide treated activated carbon. J Hazard Mat 103(1-2):141-152. [ Links ]
Marsh, H., E. A., Heintz, F., Rodríguez-Reinoso. 1997. Introduction to Carbon Technology. Capitulo 2. Universidad de Alicante, Secretariado de Publicaciones. Alicante, España. [ Links ]
Navarrete, L. F., L., Giraldo, M. C., Baquero. 2005. Activated carbon: effect of the washing with sulfuric acid of the lignocellulosic precursor, shell of african palm, on the processes of carbonization and activation. Rev Colomb Quim 34(1):67-78. [ Links ]
Norit. Norit RO 3515. 1999. Características del producto. [ Links ]
Pradhan, B. K., N. K., Sandle. 1999. Effect of different oxidizing agent treatments on the surface properties of activated carbons. Carbon 37(8):1323-1332. [ Links ]
Rajakovic, L. V., M. D., Ristic. 1996. Sorption of boric acid and borax by activated carbon impregnated with various compounds. Carbon 34(6):769-774. [ Links ]
Rodríguez-Reinoso, F., M., Molina-Sabio. 1998. Carbones activados a partir de materiales lignocelulósicos. Quibal. Química e Industria 45(9):563-571. [ Links ]
Rosas, J. M., J., Bedia, J., Rodriguez-Mirasol, T., Cordero. 2009. HEMP-derived activated carbon fibers by chemical activation with phosphoric acid. Fuel 88(1:)19-26. [ Links ]
SCFI (Secretaría de Comercio y Fomento Industrial). 1982. Norma mexicana NMX-B-158-1982, métodos para determinar el azufre total en la muestra de carbón y coque. http://200.77.231.100/work/normas/nmx/1982/nmx-b-158- 1982.pdf. [ Links ] [ Links ]
Singh, B., P., Lal. 2010. Preparation and characterization of activated carbon spheres from polystyrene sulphonate beads by steam and carbon dioxide activation. J Appl polym Sci 115(4):2409-2415. [ Links ]
Song, X., H., Liu, L., Chenga, Y., Qu. 2010. Surface modification of coconut-based activated carbon by liquid-phase oxidation and its effects on lead ion adsorption. Desalination 255(1- 3):78-83. [ Links ]
Studebaker, M. L. 1957. The Chemistry of Carbon Black and Reinforcement. Rubber Chem Technol 30:1400-1484. [ Links ]
Tongpoothorn, W., M., Sriuttha, P., Homchan, S., Chanthai, C., Ruangviriyacha. 2011. Preparation of activated carbon derived from Jatropha curcas fruit shell by simple thermochemical activation and characterization of their physicochemical properties. Chem Eng Res Des 89(3):335-340. [ Links ]
Recibido: julio 19 de 2011. Aceptado para su publicación: agosto 30 de 2011.
