










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista de la Academia Colombiana de Ciencias Exactas, Físicas y Naturales
versión impresa ISSN 0370-3908
Rev. acad. colomb. cienc. exact. fis. nat. vol.36 no.139 Bogotá abr./jun. 2012
QUÍMICA
* Este trabajo se realizó en el Centro de Investigaciones en Catálisis (CICAT) UIS, financiado por COLCIENCIAS con la participación de Carlos Alberto Páez M., Nelson Jair Castellanos M., estudiantes de doctorado en Química, Oscar Lozada estudiante de pregrado en Química, el doctor Fernando Martínez Ortega, professor Titular de la Escuela de Química de la UIS y del professor Henri Arzoumanian, professor Emérito Universidad Paul Cezanne de Marsella, Francia.
** Profesor Emérito, Universidad Industrial de Santander, Bucaramanga - Colombia, edgarpaezmozo@gmail.com
RESUMEN
En este trabajo se presenta la formación de un sistema verde bioinspirado para la Transferencia de Oxígeno (TAO). Se estudió la TAO a 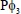 y etilbenceno empleando oxígeno molecular y luz UV-Vis, el catalizador consiste en complejos del tipo [Mo(=O)2L], L=bipiridil (bipi), bipirazolil (bpz) anclandos sobre TiO2. La TAO se estimula por una corriente electrónica, generada por acción de radiación UV-visible sobre TiO2, que es transferida a la unidad Mo=O para propiciar la TAO. La contribución del anión superóxido O2-, formado por reacción de los e- fotogenerados con el O2, parece jugar un papel importante en la regeneración del centro activo Mo(=O)2 ya que en ausencia de luz la TAO solo es estequiométrica.
y etilbenceno empleando oxígeno molecular y luz UV-Vis, el catalizador consiste en complejos del tipo [Mo(=O)2L], L=bipiridil (bipi), bipirazolil (bpz) anclandos sobre TiO2. La TAO se estimula por una corriente electrónica, generada por acción de radiación UV-visible sobre TiO2, que es transferida a la unidad Mo=O para propiciar la TAO. La contribución del anión superóxido O2-, formado por reacción de los e- fotogenerados con el O2, parece jugar un papel importante en la regeneración del centro activo Mo(=O)2 ya que en ausencia de luz la TAO solo es estequiométrica.
Se observó que aunque el complejo bipirazolil es un buen agente en la TAO, debido a su propiedad donadora de electrones, el sistema es pobre en la activación de Oxígeno debido a su labilidad a la lixiviación en medio orgánico, para este efecto se obtiene un mejor balance con el ligando Bipi. Con estos sistemas se abre una perspectiva para el desarrollo de procesos verdes para la oxidación selectiva de compuestos orgánicos.
Palabras clave: Dioxigeno, Complejos dioxoMolibdeno, Transferencia Atómos de Oxígeno, Catalizadores Anclados sobre Titania.
ABSTRACT
In this paper, the formation of an Oxygen Transfer (OAT) green bioinspired system was developed. The OAT to and ethylbenzene, using molecular oxygen and UV-Vis light is reported. The catalyst consists of complexes of the type [Mo(=O)2L], L=bipyriyll(bipi), lbipyrazolyl(bpz) bound to TiO2. The OAT is stimulated by an electron current, generated by the action of UV-visible light on TiO2, which is transferred to the Mo = O unit to promote the OAT. The contribution of the superoxide anion O2-, formed by the action of the e-photogenerated with O2, seems to play an important role in regenerating the Mo(= O)2 active center since in the absence of light OAT is stoichiometric.
Although the bipirazolyl is a good OAT agent due to its electron donating property, the system is poor in the activation of oxygen due to its organic media leaching lability, with the ligand Bipi a better balance is obtained. These systems offer a good opportunity to develop green processes for selective oxidation of organic compounds.
Key words: Dyoxygen, DioxoMolibdenum Complexes, Oxyen Atom Transfer, Titania Anchored Catalysts.
Introducción
La formación directa de compuestos orgánicos oxigenados, empleando oxígeno molecular, reviste una importancia enorme pues es la esencia misma de numerosas transformaciones biológicas y químicas. La transferencia de oxígeno hacia un compuesto orgánico es una operación muy delicada, que en la naturaleza es realizada en condiciones ambientales de temperatura, presión y de manera muy selectiva por las enzimas (oxotransferasas e hidroxilasas) [1,2].
En el proceso de transferencia de O, aquellas enzimas que tienen un centro mononuclear de molibdeno en su sitio activo, son de particular interés. Las oxotransferasas de molibdeno, durante el proceso TAO su centro activo alterna entre la unidad Mo(VI)O2 y el Mo(IV)O [3], lo mismo se ha observado con catalizadores bioinspirados con complejos sencillos de Mo [4,5]. Por lo anterior se hace muy importante estudiar los factores que facilitan la donación selectiva del oxígeno.
Se ha observado que la transferencia de átomo de Oxígeno (TAO) en solución, se facilita tanto electrónica como energéticamente por la presencia de un grupo oxo "espectador vecino" [6] capaz de donar carga electrónica al centro activo, es decir de transferir densidad electrónica a la unidad Mo=O. La transferencia electrónica (TE) hacia el centro activo del donor de Oxígeno es un factor clave en los procesos de TAO.
Cuando se emplea O2 en los procesos de TAO se requiere de la activación reductiva del oxígeno, para lo cual es necesaria la presencia de electrones y de H+. En un sistema bioinspirado una corriente de electrones puede generarse, entre otros medios, a partir de un Donor foto-excitado como: TiO2, Porfirinas, Ftalocianinas u otras moléculas con niveles de energía accesibles y apropiados para favorecer termodinámicamente la Transferencia de Electrones hacia un Aceptor.
En este trabajo se describe la formación de un sistema bioinspirado conformado por complejos de MoO2L (L = Bipiridil, bipirazolil) anclados sobre TiO2 que en presencia de luz UV-Vis, permite la transferencia de oxígeno hacia moléculas orgánicas, empleando O2 en condiciones ambientales.
La transferencia de oxígeno
En las oxotransferasas y en algunos catalizadores sintéticos de Mo (y W), se ha mostrado que la unidad MoVI(=O) es la responsable de la Transferencia de átomo de Oxígeno. Se ha propuesto que la TAO en solución tiene lugar después de un ataque nucleofílico de un sustrato (como la tri-fenilfosfina, :) sobre el orbital π* de uno de los grupos Mo=O reduciendo el metal [3, 7-9]. Se cree que la fosfina se aproxima a la unidad MoO2 con un ángulo de 90º, maximizando la superposición orbital entre el par solitario de la fosfina y el orbital π* del grupo Mo=O, seguido de una rotación alrededor del enlace Mo-O. Esta rotación permite la formación de un enlace  disminuyendo a 1 el orden del enlace del Mo=O. Al mismo tiempo el enlace Mo=O restante (vecino) se hace más fuerte, debido a la retro-donación π hacia el metal, aumentando el orden de enlace a 3, con lo cual el proceso en general se hace termodinámicamente favorable, tanto que la pérdida de una unidad de Mo=O (estable) se compensa con el reforzamiento del enlace que permanece. Este efecto oxo espectador juega un papel importante para determinar la reactividad química de los centros MoO2.
disminuyendo a 1 el orden del enlace del Mo=O. Al mismo tiempo el enlace Mo=O restante (vecino) se hace más fuerte, debido a la retro-donación π hacia el metal, aumentando el orden de enlace a 3, con lo cual el proceso en general se hace termodinámicamente favorable, tanto que la pérdida de una unidad de Mo=O (estable) se compensa con el reforzamiento del enlace que permanece. Este efecto oxo espectador juega un papel importante para determinar la reactividad química de los centros MoO2.
La transferencia electrónica del enlace π* Mo=O al centro metálico debilita el enlace O-Mo, que al romperse forma las especies Mo(IV) y O [6]. Se han realizado ajustes termodinámicos de los centros de Mo para promover reacciones de TAO en enzimas, mostrando que la TAO depende de factores que estabilizan o desestabilizan los estados de oxidación Mo(IV) y Mo(VI) [10].
Los procesos de TAO en la naturaleza requieren de la activación reductiva del oxígeno, para lo cual se necesita del concurso de un sistema capaz de producir una corriente de electrones y de protones, función que es realizada por la NaDPH. La TE hacia el centro activo del donor de Oxígeno es clave en el proceso de TAO. En el estudio de modelos enzimáticos artificiales, se ha encontrado que para lograr la activación del oxígeno molecular, se requiere del suministro de electrones desde una fuente externa: como la descomposición catalítica de Hidrógeno, el concurso de compuestos reductores o un proceso foto-estimulado.
Transferencia electrónica foto-estimulada
Se puede lograr la Transferencia de Electrones (TE) desde una molécula en un estado fotoexcitado, a otra molécula en su estado basal. Este proceso depende del cambio de la energía libre total  GTE y de la energía o barrera de activación en la transferencia.
GTE y de la energía o barrera de activación en la transferencia.
Se pueden distinguir dos casos para la TE:
1. En el que los reaccionantes están disueltos, son móviles y libres para aproximarse a distancias cercanas, dentro del periodo de vida del Donor excitado, para formar pares iónicos [11]. El Donor y los reaccionantes tienen libertad para difundirse hasta una distancia apropiada para formar el llamado complejo de encuentro (en solución) y
2. en el que existen factores estructurales que mantienen al donor (D) y el aceptor (A) separados por un puente (b) a distancias fijas. Durante el tiempo de vida del Donor excitado, la TE es permitida, si los factores nucleares son favorables. La energética y la cinética de la transferencia dependen de: la estructura de los reaccionantes, de la distancia entre ellos, de la naturaleza y polaridad del medio y de los efectos coulómbicos. El cambio en la energía libre de Gibbs (GTE<0) se puede calcular empleando la ecuación de Rehm-Weller, basada en los potenciales redox del donor: D*, Aceptor: A y el puente: b [12]).
Donación de oxígeno foto-estimulada (TAOF)
En el proceso de TAO Foto-Estimulada se pueden distinguir varias etapas:
1. Interacción del Sustrato con el Complejo Donor de Oxígeno (ataque nucleofílico),
2. transferencia electrónica desde un donor de electrones foto-excitado (D*) hacia el Complejo "donor de Oxígeno" (A),
3. proceso de transferencia de oxígeno y
4. recuperación del centro activo (Mo=O).
Se conocen numerosos casos de TAO con complejos de MoO2Ln (Ln = Ligando voluminoso), que han facilitado el estudio desde el punto de vista termodinámico [13-15]. Arzoumanian ha observado que el complejo MoVI(=O)2(LNS2) es capaz de oxidar estequiométricamente a la Ph3P en DMF [16] para producir Ph3PO, reduciéndose a MoIV(=O)(LNS2). Luego el complejo vuelve a su estado inicial por interacción con un solvente donor de O, como el dimetil sulfóxido (esquema 1).
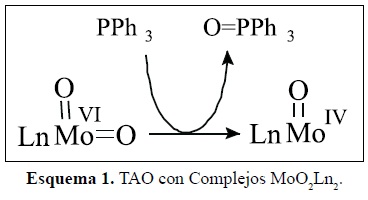
Se observa que a pesar de la estabilidad del enlace Mo=O (H=-45 Kcal/mol), la reactividad del grupo Mo (=O)2 permite que el enlace en solución sea relativamente lábil y bajo las condiciones apropiadas realiza la oxotransferencia. Por otra parte, termodinámicamente se ha estudiado una gran variedad de oxodonores y oxoaceptores, determinándose por ejemplo como buenos oxodonores los grupos sulfóxido, nitratos y N-óxidos, y como buenos oxoaceptores: fosfinas y sulfitos.
En la oxotransferencia desde el centro MoVIO2, hacia el oxo-aceptor, el mecanismo propuesto involucra un ataque nucleofílico por el par de electrones solitarios de la fosfina sobre el orbital π* de uno de los enlaces Mo=O, llevando a la correspondiente reducción, seguido por el desplazamiento del producto, como se muestra en el esquema 2 [17,18].

Se ha propuesto que el substrato se coordina con uno de los grupos Mo=O, formando un intermediario bidentado luego del ataque nucleofílico, logrando la formación de un "complejo fosfatado" de MoV, cuyas señales EPR son similares a las observadas en la sulfito oxidasa [19].
En procesos de Transferencia Electrónica Fotoinducida (TEF) como ocurre en sistemas amino-carbonilo, se ha establecido que existe un par electrónico nN (no enlazante sobre el N) del grupo amino (Donor) en un nivel energético más alto que el estado basal nO (nivel no enlazante sobre el oxígeno) del C=O (Aceptor), se ha mostrado que la TE exoenergética solo puede ocurrir una vez que se haya producido la excitación n0→π*(C=O) ver Figura 1. Lo anterior también se cumple para donores electrónicos menos activos como los alquil- carboxilatos, hidrocarburos insaturados, aromáticos y otros. [20] .
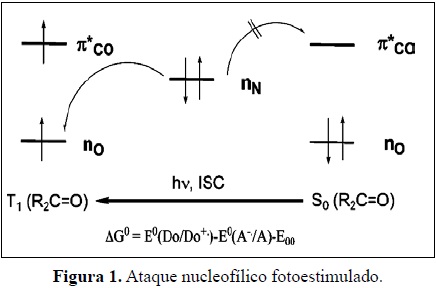
La energía libre de la transferencia se expresa por medio de:
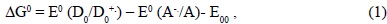
E0 son los potenciales redox y E00 es la energía electrónica 00 del estado excitado del aceptor en este caso E00 = hn [n→π*(C=O)] [11].
H. Arzoumanian [16] estudiando reacciones de TAO con los complejos de molibdeno observó un fuerte aumento en la TAO del Mo(VI)Bipi (SCN)2(O)2 hacia una molécula orgánica en DMSO, cuando empleó radiación UV de l = 350 nm. Esto se puede interpretar, de acuerdo con la discusión anterior, debido a una disminución del orden de enlace del Mo=O como consecuencia de una transferencia electrónica foto inducida: 1- nO + h→*p del Mo=O: que deja al nivel n0 semi lleno, 2- TE del par solitario de la Fosfina (:P) hacia el nivel nO del Mo=O: facilitando así la donación de oxígeno.
Tranferencia electrónica a través de un puente
Si el Donor de electrones y el Aceptor están unidos por un puente ó separador molecular (b), se pueden tomar como una SUPRAMOLÉCULA, que experimenta deformaciones de alta energía en los enlaces y de baja energía con los dipolos del solvente. La TE puede tomarse como una transición no radiativa entre dos superficies de energía potencial, caracterizada por la constante de velocidad kET ec. (1) [11,25].
Puesto que la TE juega un papel muy importante en los procesos de TAO haremos algunas consideraciones sobre ciertos factores que afectan la TE hacia el centro donor de oxígeno través de un puente molecular.
La velocidad de la Transferencia Electrónica puede expresarse según el formalismo de Marcus-Hush-Sutin [22,23]:
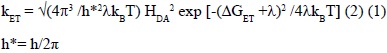
Donde λ es la barrera intrínseca (ó energía reorganizacional) y HDA es el término de acoplamiento electrónico (o la energía de estabilización por resonancia), entre el estado inicial y final [24].
En los procesos de TE en un sistema D-b-A, la energética y la distancia entre D y A juegan un papel importante en el mecanismo. Se han logrado evidencias experimentales del papel que desempeña la barrera energética o brecha energética del túnel entre los portadores de carga. La TE a través de un túnel del D-b-A depende en gran parte de HDA2, que es > 0 aun para longitudes grandes del puente (dp). [26].
El factor pre exponencial de la ec. (2) depende de la energía del puente y de la energía del donor. Este factor es grande, si EDA es grande y varía muy ligeramente con la altura de la barrera EDb. Si EDb es pequeño la TE varía rápidamente con d, el factor exponencial se aproxima a 1[29,30].
La transferencia se puede interpretar dentro del marco de la teoría del "superintercambio", que describe el acoplamiento electrónico entre D-b-A a distancias grandes, en un sistema en el que D y A están separados por un puente con n unidades idénticas. El acoplamiento electrónico HDA depende de tres acoplamientos electrónicos figura (2) que son: el acoplamiento entre D y la primera unidad del puente, el acoplamiento entre las unidades del puente y el acoplamiento entre la última unidad y A. HDA es inversamente proporcional a ε que es la brecha energética del túnel, que es la diferencia energética entre el sistema D-b-A y la energía de los estados localizados en el puente (en el estado de transición). En términos prácticos la brecha ε se puede relacionar con los potenciales redox de D, b, A y se conoce como energía de inyección;
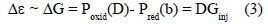
Como lo sugiere la ec. (3), Ginj se puede determinar a partir de los potenciales de oxido-reducción por voltametría y espectroscopía UV-Vis.
HDA disminuye exponencialmente con d y con una constante de decaimiento β (pendiente), que es directamente proporcional a ε [31,32], y puede expresarse por la ec. (4), (Fig. 2). [31,33-35].
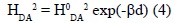
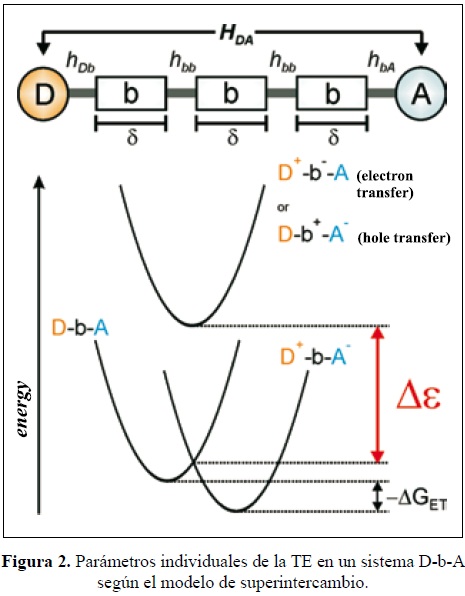
Se pueden identificar varios factores que influyen en la TE por un puente:
TE en función de la brecha del puente Ginj
En puentes D-b-A de diferente longitud (db) se pueden obtener curvas de kTE vs db con pendientes βi para diferentes valores de Ginj (que es aproximadamente la diferencia entre el potencial de oxidación de D y el potencial de reducción del puente) de acuerdo a la teoría de Superintercambio [24]. La experiencia muestra que la transferencia electrónica se hace mayor si Ginj se es menor y generalmente este factor tiene mayor peso que la distancia entre D y A [26].
La TE entre un donor: Zn(II)Pn (D) y un aceptor Au(III) Pn (A), (Pn: porfirina), separados por una distancia de 25 A y conectados por un puente formado por oligo-p-fenilen etinileno y un espaciador intermedio: benceno, naftaleno y antraceno. Se observó que la TE entre D y A, en diferentes
solventes, la velocidad aumenta al ir del benceno al naftaleno y al antraceno, como unidad central del puente, (de 2.2 a 4.3 y a 9.1 x 109 s-1) basándose en datos experimentales de espectroscopía de tiempo resuelto y de estado estacionario (steady-state), se pueden obtener valores de HDA: 5.5-7.5 cm-1 para el benceno, 7-10 cm-1 para el naftaleno y 11-17 cm-1 para el antraceno [36].
La brecha energética del túnel, se asoció en este caso, con las diferencias de energía entre, la banda de absorción más baja del puente y la banda de absorción más baja del donor (transición HOMO-LUMO), observadas por espectroscopía UV-Vis. Los valores determinados para inj son: 1.44 eV (benceno), 1.07 eV (naftaleno) y 0.48 eV (antraceno). Como puede observarse la TE es mayor entre mayor sea la deslocalización p del puente.
TAOF por Complejos Mo(VI)O2Ln/TiO2
En nuestro laboratorio hemos observado que compuestos del tipo MoVI(=O)2L (5) anclados sobre TiO2: ([MoVI(=O)2L]/ TiO2) (6) Figura 3, son capaces de realizar la TAO (hacia: y algunos compuestos alquil aromáticos) en presencia de luz (UV y Vis) y O2 en condiciones ambientales. Este resultado se ha asociado al efecto de la TEF desde la Banda de Conducción del TiO2 hacia la unidad π* del Mo=O [37-40].
Cuando D (TiO2) y A [MoVI=O] están unidos mediante un puente molecular (b= carboxilato-Ligando: Bipidil y bipirazolil), se puede considerar que el proceso de TEF es "intramolecular" y depende principalmente de la energía de inyección, que es la brecha energética entre D y b: G = Poxid(D)- Pred(b) = Ginj y de la distancia entre D y A. Las interacciones orbitales pueden ocurrir a través de los enlaces, el acoplamiento decrece exponencialmente con el número de enlaces pues los electrones del Donor pasan a los orbitales LUMO de las moléculas del puente y luego al Aceptor. El mecanismo de la TE depende de la brecha de energía entre los orbitales del D y A y de los orbitales del puente [11].
Nuestros experimentos indican que es posible construir un sistema bioinspirado, anclando un complejo Mo(=O)2Ln sobre un Donor (D = TiO2), [Mo(=O)2Ln/D] que en presencia de radiación UV-visible puede generar una corriente electrónica foto-inducida sobre la unidad Mo=O que propicia la TAO.
La transferencia catalítica de O se examinó empleando el sistema (6) Figura 3, donde D = TiO2 y A = [MoV(=O)2Bipi], usando trifenilfosfina y varios arilalcanos como receptores de O y empleando O2 directamente como el donor de oxígeno.
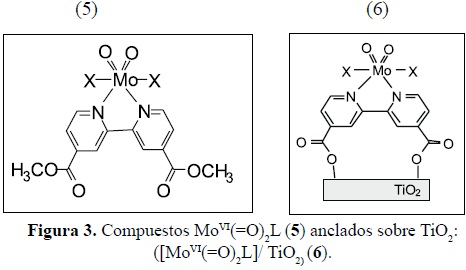
La oxidación de la PPh3 con el [MoV(=O)2Bipi] libre (5), tanto en la oscuridad como en presencia de luz, presenta una transferencia estequiométrica de oxígeno. El mismo resultado se obtuvo con una mezcla mecánica de [MoV(=O)2Bipi] libre (5) y TiO2. Por otro lado el sistema formado por el complejo de Mo anclado sobre TiO2 : [MoV(=O)2L]/ TiO2 (6) mostró en la oscuridad una menor transferencia que el complejo libre (5), pero en presencia de luz UV-Vis mostró un incremento en su actividad catalítica 3 a 4 veces mayor [38,39].
Estos resultados sugieren un efecto sinérgico debido a la fotogeneración del flujo electrónico desde la matriz semiconductora hacia la esfera de coordinación del Mo. Cuando se realizó el experimento en ausencia de oxígeno, después de evacuar el aire e inyectar N2, la oxidación de PPh3 corresponde a la reducción estequiométrica del Mo (VI) Dioxo presente en el medio de reacción. Al reintroducir el O2 retorna la actividad catalítica. Una tendencia similar se observó en la TAO hacia etilbenceno, cumeno y tetralina [38,39]. Los posibles caminos para la TE y los valores relativos de los niveles de energía se indican en el esquema 3.
Los valores para la banda de conducción y la banda de valencia del TiO2 son -0.7 V y 2.5 V vs SCE respectivamente [41,42], mientras que los valores asignados para los orbitales HOMO y LUMO se obtienen de los potenciales de oxidación- reducción y de espectroscopía UV-Vis en solución, medidos en nuestro laboratorio.
El camino 3 (esquema 3) es posible en presencia de radiación de ~350 nm que corresponde a la transferencia n →*π del Mo=O.
Cuando se repiten los mismos experimentos empleando el complejo con ligando bipirazolil (bpz) (7) y (bipi)(8), inicialmente la Oxo-transferencia es más alta para el complejo bipirazolil que para el bipiridil por un factor cercano a tres. Este resultado sigue la tendencia observada en la oxidación de fosfinas con TBHP, con un complejo heteroescorpionato de MoO2 [43] análogo a (7).
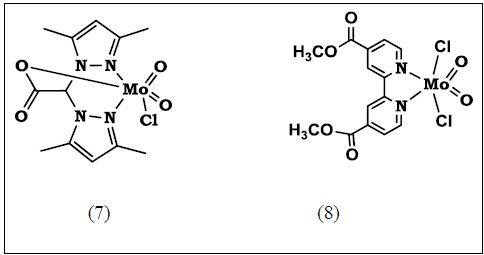
Hemos podido observar que aunque inicialmente la TAO con bpz es más rápida que con la bipi, el complejo bpz se lixivia perdiendo así su actividad.
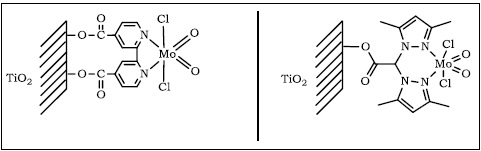
Conclusiones
Es posible construir un sistema bioinspirado para la Transferencia de Oxígeno a un sustrato empleando oxígeno molecular, anclando complejos del tipo Mo(=O)2Ln sobre un Donor (D = TiO2), [Mo(=O)2Ln/D] que en presencia de radiación UV-visible puede generar una corriente de electrones sobre la unidad Mo=O para propiciar la TAO, con lo cual se hace una contribución importante en el desarrollo de procesos de Química verde. La participación del ataque nucleofílico foto estimulado del sustrato: (en presencia de la radiación apropiada para efectuar la transición n →*p del Mo=O ( 350 nm) para facilitar la TE de la: (D) hacia el Mo=O (A*) puede ser de importancia como se sugiere en el esquema 4 (camino 3). La contribución del anión superóxido O2-, formado por reacción de los e- fotogenerados con el O2, parece jugar un papel importante en la regeneración del centro activo (Mo=O2) ya que en ausencia de luz la TAO es estequiométrica (no es catalítica). La contribución del anión superóxido O2-, formado por reacción de los e- fotogenerados con el O2, parece jugar un papel importante en la regeneración del centro activo (Mo=O2) ya que en ausencia de luz la TAO solo es estequiométrica
Aunque el complejo bipirazolil es un buen agente en la TAO debido a su propiedad donadora de electrones, el sistema es pobre en la activación de Oxígeno debido a su labilidad a la lixiviación en medio orgánico, para este efecto se obtiene un mejor balance con el ligando Bipi.
Bibliografía
Majumbar A.,Sarkar S., Biomimetic chemistry of molybdenum and tungsteno enzymes: A structural-functionsl modeling approach, Coord. Chem. Revs 255(2011)1039-1054 [ Links ]
Holm R., Soloman E., Majundar A., Tenderholt A., Comparative molecular chemistry of molybdenum and tungten and its relation to hydroxylase and oxotransferase enzymes, Coord. Chem. Revs., 235 (2011)993-1015 [ Links ]
R. Hille et al., Mechanistic aspects of molibdenum-containing enzymes, FEMS Microbiology Reviews 22(1999)489-501 [ Links ]
Holm R., The biologically relevant oxygen atom transfer chemistry of molybdenum: from synthetic analogue Systems to enzymes. Coord. Chem. Revs. 100 (1990)183-221 [ Links ]
Enmark J., Young C., Bioinorganic chemistry of pterin-containing molybdenum and tungten enzymes. Adv. Inorg. Chem. 40(1993)1-88 [ Links ]
Young C., Biomimetic Chemistry of Molybdenum, ch.9 p413, Biomimetic Oxidations Catalyzed by Transition Metal Complexes, Editor Bernard Meunier, Imperial College Press 2000 [ Links ]
Keith J.M., et al. Oxygen atom transfer calaysis: Ligand effects on the key reaction barrier in molybdenum (VI) dioxo systems, J. Mol. Cat.,A: Chem 324(2010)15-23 [ Links ]
Biomimetic Oxidations Catalyzed by Transition Metal Complexes, Editor Bernard Meunier, Imperial College Press 2000 [ Links ]
Pietsch, M.A. and Hall, M.B., Theoretical Studies of the oxo- transfer reaction of dioxomolibdenum enzymes, 35,(1996)1273-1278 [ Links ]
R. H. Holm and J. M. Berg, Acc. Chem. Res., 1986, 19, 363-370 [ Links ]
Kavarnos G., J., Fundamental Concepts of Photoinduced Electron Transfer, Topics in Current Chemistry, 156(1990)21-58 [ Links ]
Griesbeck A., Hoffmann N., Warzecha K., Acc. Chem. Res. 40(2007)128- 140 [ Links ]
Hille, R., Chem. Rev. 1996,96,2757-28116 [ Links ]
Arzoumanian H. et al., Inorg. Chem. 1994, 33, 3177- 3179 [ Links ]
Arzoumanian H. et al., New J. Chem. 1996,20, 699-705 [ Links ]
Arzumanian H., et. al., Thiocyanatodioxomolybdenum (VI) complexes as efficient oxidaizing agents, J. Mol. Cat. A: Chem. 117, (1997),471- 478 [ Links ]
M. Minelli, K. Yamanouci, J.H. Enemark, P. Subraminian, B.B. Kaul, J.T. Spence. Inorg. Chem., 13 (1984) 2554 [ Links ]
N. Teruel, A. Sierralta, J. Mol. Cat. Cat. A.: Chem., 107 (1996) 379 [ Links ]
Gray, R. C., Quart. Rev. Biophy. 1988, 21: 299-32 [ Links ]
Griesbeck, Acc. Chem. Res., 40,2(2007)128-140 [ Links ]
Gray, H. B., Winkler, J. R., Long-range electron transfer, Proc. Natl. Acad. Sci. USA. 2005,102,3534-3539 [ Links ]
Marcus, R. A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta 1985, 811, 265–322 [ Links ]
Kochi et al, JACS125,9,2003,2559-2571 [ Links ]
Rosokha S., V.; Kochi A., Y., K., Fresh Look at Electron-Transfer Mechanisms via the Donor/Acceptor Bindings in the Critical Encounter Complex, Acc. Chem. Res., 41,5(2008)6421-653 [ Links ]
Marcus, R. A., Sutin, N., electron transfer in chemistry and biology. Biochim. Biophys. Acta 811,(1985)265-322 [ Links ]
Wenger O., How Donor-Brdge-Acceptor Energetics influence Electron Tunneling Dynamics and their Distance Dependences, Acc. Chem. Res. (2011),44,1,p25-35 [ Links ]
B. Abinsson, M. P. Eng, K. Pettersson and M. U. Winters, Electron and energy transfer in donor-acceptor Systems conjugated molecular bridges Phys. Chem. Chem. Phys., 2007,9,5847-5864,DOI: 10.1039/ b706122f [ Links ]
Gray, H.B., Windler, J.R., Long-range electron transfer. Proc. Natl. Acad. Sci. USA 102,(2005),3534-3539 [ Links ]
Jortner, J., et. al., Charge transfer and transport in DNA. Proc. Natl. Acad. Sci. U:S:A. 1998, 106, 12759-12765 [ Links ]
Tong, G. S., et. al. Tunneling energy effects on GC oxidation in DNA. J. Phys. Chem. B 2002, 106, 2381-2392 [ Links ]
McConnell, H.M. Intramolecular charge transfer in aromatic free radicals. J. Chem. Phys. 27,(1961),508-515 [ Links ]
Paddon-Row, M. N. Investigating ion-range electron-transfer processes with rigid, covalently-linked donor-(nobrornylogous bridge)-acceptor Systems. Acc. Chem. Res. 27(1994),18-25 [ Links ]
Y. A. Berlin, A. L Burin and P. Rempala, Chem. Phys., 2002,275, 61-74 [ Links ]
Y. A. Berlin, G. R. Hutchison, P. Rempala and M. A. Ratner, Phys. Rev. A, 2003,107, 3970-3980 [ Links ]
M. U. Winters, K. Petersson, J. Marternsson and B. Albinsson, Chem.- Eur. J., 2005,11,562-573 [ Links ]
Kilsá K., Kajanus J., Macpherson A. N., Márienson J., Albinson B., Bridge-dependent electron transfer in porphyrin-based donor-bridgeacceptor Systems. J. Am. Chem. Soc. 2001,123,3069-3080 [ Links ]
Kilsá K., Kajanus J., Macpherson A. N., Márienson J., Albinson B., Bridge-dependent electron transfer in porphyrin-based donor-bridgeacceptor Systems. J. Am. Chem. Soc. 2001,123,3069-3080 [ Links ]
Carlos A. Páez, Nelson J. Castellanos, Fernando Martínez O., Fabio Ziarelli,Giuseppe Agrifoglio, Edgar A. Páez-Mozo, Henri Arzoumanian, Oxygen atom transfer photocatalyzed by molybdenum(VI) dioxodibromo-(4,4'-dicarboxylate-2,2-bipyridine) anchored on TiO2, CatalysisToday133–135(2008)619–624. [ Links ]
Carlos A. Páez, Oscar Lozada, Nelson J. Castellanos, Fernando Martínez O., Fabio Ziarelli, Giuseppe Agrifoglio, Edgar A. Páez-Mozo, Henri Arzoumanian, Arylalkane photo-oxidation under visible light and O2 catalyzed by molybdenum(VI) dioxo-dibromo(4,4-dicarboxylato- 2,2-bipyridine) anchored on TiO2, Journal of Molecular Catalysis A: Chemical 299 (2009) 53–59 [ Links ]
Henri Arzoumanian, Nelson J. Castellanos, Fernando Martínez O., Edgar A. Páez-Mozo, and Fabio Ziarelli, Silicon-Assisted Direct Covalent Grafting on Metal Oxide Surfaces: Synthesis and Characterization of Carboxylate N,N-Ligands on TiO2, European Journal of Inorganic Chemistry, 2010, DOI: 10.1002/ejic.200901092 [ Links ]
Galoppini E., Coord. Chem. Rev. 2004, 248,1283 [ Links ]
Kopidakis, A. J. F., De Lagemaat J. V., Coord. Chem. Revs. 2004, 248, 1165 [ Links ]
Hammes B.S., Chohan B.S., Hoffman J.T., Einwachter S., Carrano C.J., Inorg. Chem. 2004, 43, 7800 [ Links ]
Recibido: 2 de abril de 2012
Aceptado para publicación: 4 de junio de 2012
