










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Academia Colombiana de Ciencias Exactas, Físicas y Naturales
Print version ISSN 0370-3908
Rev. acad. colomb. cienc. exact. fis. nat. vol.39 no.151 Bogotá Apr./June 2015
https://doi.org/10.18257/raccefyn.183
doi: http://dx.doi.org/10.18257/raccefyn.183
Laboratorio de Biología Molecular y Patogénesis, Departamento de Ciencias Fisiológicas, Escuela de Ciencias Básicas, Facultad de Salud, Universidad del Valle, Campus de San Fernando, Cali, Colombia. *Correspondencia: Felipe García Vallejo, labiomol@gmail.com
Recibido: 14 de enero de 2015. Aceptado: 6 de abril de 2014
Resumen
La selección de los sitios de integración del ADN retroviral en el genoma es crucial para moldear la dinámica de la infección. El objetivo de este estudio fue analizar la combinación específica de las características genómicas de la célula infectada que condicionarían la integración simultánea de ambos retrovirus. A partir de 203 secuencias de ADN humano vecinas a las repeticiones terminales largas (long terminal repeat, LTR) de ambos virus, depositadas en el GenBank, y mediante distintas herramientas computacionales, se hizo una simulación bioinformática para determinar la integración del VIH y el -HTLV-1 en una extensión de 100 kb, así como la localización cromosómica del provirus, el número de genes, su proceso molecular y función asociada, las islas CpG, las secuencias Alu y los elementos nucleares dispersos largos (long interspersed nuclear element, LINE), y su expresión en poblaciones de linfocitos de los genes blanco de la integración. El 47,3 % de las integraciones de ambos virus se localizó en regiones ricas en elementos repetidos. La integración en los genes de la clase II ocurrió en los intrones (p<0,05). Se observó una distribución cromosómica diferencial de ambos provirus en la que el HTLV-1 se localizó en regiones pericentroméricas y centroméricas, mientras que el VIH-1 lo hizo en zonas teloméricas y subteloméricas (p<0,001). El ambiente común para la integración de los linfocitos en el genoma estuvo conformado por genes codificantes de proteínas de unión a moléculas y de transducción de señales, así como por un elevado número de islas CpG y de repeticiones Alu. A partir de la simulación bioinformática de la integración entre el VIH-1 y el HTLV-1, se aportó evidencia que sustenta la hipótesis de que una combinación específica de variables genómicas condicionaría el proceso de integración diferencial simultánea de ambos retrovirus.
Palabras clave: retrovirus, integración viral, linfocitos, islas de CpG, genes clase II, simulación por computador.
Abstract
The selection of retroviral cDNA integration sites in the human genome is a critical step to condition the dynamics of infection. The objective was to analyze the combination of genomic characteristics of infected cells that would condition the genomic profiles of HIV-1/HTLV-1 simultaneous integration. We carried out a computer simulation using 203 human genome sequences flanking 3'LTR of both retroviruses previously deposited in the GenBank, and applying several computational tools. The analyses were focused on determining the chromosomal integration, CpG islands, Alu sequences and the expression of integration target class II genes in lymphocytic populations in a 100 kb chromatin structure associated with simultaneous integration. We found 47.3% of simultaneous cDNA integrations localized in regions rich in repetitive elements. The rest of retroviral cDNA integrations occurred in class II gene introns (p<0.05). We determined a differential chromosomal distribution for both types of provirus where HTLV-1 provirus were preferentially placed in pericentromeric and centromeric regions in contrast with HIV-1 distribution, which was registered in telomeric and subtelomeric zones (p<0.001). The genomic environment of integration for both retroviruses was characterized by genes encoding molecular binding and signal transduction, as well as by high density of CpG islands and Alu sequences. The data resulting from the computer simulation did support the hypothesis that a combination of specific chromatin characteristics would determine the dynamics of HIV-1/HTLV-1 simultaneous integration process.
Key words: Retrovirus, provirus integration, lymphocytes, CpG island, class II genes, computer simulation.
Introducción
Las infecciones concomitantes por el virus de la inmuno-deficiencia humana tipo 1 (VIH-1) y el virus linfotrópico humano tipo 1 (HTLV-1) constituyen problemas importantes de salud en aquellas zonas donde puede ocurrir la transmisión simultánea de ambos retrovirus (Casoli, et al., 2007; Laurentino, et al., 2005; Góngora-Bianchi, et al., 2003; Brites, et al., 1997). A pesar de los reportes sobre la infección concomitante en diferentes países, en Colombia no se han registrado oficialmente casos de infección simultánea con los dos virus en la costa del Pacífico, que es un área endémica para su circulación. Aunque la infección concomitante con ambos retrovirus se ha descrito epidemiológicamente en otros países, todavía existe controversia en torno a cuál es el efecto de la interferencia con la función inmunológica del paciente y el pronóstico de las enfermedades asociadas (Pilotti, et al., 2013; Brites, et al., 2011; Pedroso, et al., 2011; Moriuchi, et al., 1998; Lefrère, et al., 1990).
La integración del ADN complementario (ADNc) viral en el genoma de las células infectadas es un paso necesario para la replicación del virus (Sierra, et al., 2005; Hindmarsh & Leis, 1999). Así pues, la selección de los sitios aceptores para la integración en las células hospederas se convierte en un evento crucial tanto para el retrovirus como para el hospedero (Van Maele, et al., 2006; Bushman, et al., 2005). Este proceso favorece la expresión regulada de los genes virales (Derse, et al., 2007; Mitchell, et al., 2004). En el hospedero la integración puede causar efectos adversos como la activación de protooncogenes o la inactivación de genes celulares que alteran la homeostasis celular (Nienhuis, et al., 2006). En este sentido, se ha reportado la activación de la inserción de oncogenes en varios casos de tratamiento con terapia génica (Biasco, et al., 2012; Daniel, et al., 2008). En uno de ellos se registró la integración de un vector retroviral terapéutico cerca del protooncogen LMO2, lo que contribuyó a la 'malignización' de las células T de varios pacientes sometidos a terapia génica para la deficiencia en la adenosina desaminasa (ADA) (Hacein-Bey-Abina, et al., 2003).
Recientemente se ha informado que, aunque la mayor parte del genoma hospedero es potencialmente accesible para la integración retroviral, la selección del sitio blanco para dicho proceso no se da totalmente al azar (Debyser, et al., 2015; Ambrosi, et al., 2008). En este sentido, la disponibilidad de la información sobre la secuencia completa del genoma humano ha permitido hacer estudios a gran escala para la identificación de sitios de inserción retroviral (Derse, et al., 2007; Bushman, et al., 2005). Un hallazgo importante de estos estudios es que los retrovirus de distintos géneros tienen diferentes estilos de integración, principalmente en las zonas con actividad transcripcional, en las islas CpG y en las secuencias Alu (Soto & García Vallejo, 2011; Salcedo- Cifuentes, et al., 2011; Crise, et al., 2006; Lewinski, et al., 2006)
Actualmente la mayoría de los estudios sobre la infección concomitante con VIH-1 y HTLV-1 se han centrado en sus efectos sobre la carga proviral y la viral, la expresión de los genes virales y en la posible aceleración de la enfermedad (Pedroso, et al., 2011; Casoli, et al., 2007; Laurentino, et al., 2005). Sin embargo, hasta el momento no se han analizado las características de las regiones del genoma humano donde ocurre la integración simultánea de los dos retrovirus y sus potenciales efectos en el desequilibrio de la homeostasis de la célula infectada. En este trabajo se hizo una simulación bioinformática para conocer más detalladamente las características de las regiones del genoma donde ocurren con bastantea frecuencia integraciones simultáneas de VIH-1 y HTLV-1; además, se simuló su efecto en la expresión de los genes de la clase II que han sido blanco de integración en diferentes poblaciones de linfocitos. Como hipótesis de trabajo se propuso que diferentes variables genómicas estructurales y funcionales condicionarían un comportamiento diferencial en la expresión global, el cual alteraría la homeostasis de las poblaciones de los linfocitos T-CD4+ y T-CD8+ en humanos infectados.
Materiales y métodos
Obtención de secuencias del genoma humano adyacentes a los provirus del VIH-1 y el HTLV-1
De las 464 secuencias del genoma humano que flanquean los provirus del VIH-1 de las células T CD4+ obtenidas de pacientes naturalmente infectados y reportadas por Ikeda, et al. (2007), se tomaron 130 (28 %) de la base de datos del GenBank (http://www.ncbi.nlm.nih.gov/genbank), utilizando la función ALEATORIO.ENTRE del programa Microsoft Excel. Los códigos de identificación de estas secuencias en el GenBank son los siguientes: AB256169, AB25616, AB256164, AB256161, AB256156, AB256151, AB256148, AB256144, AB256135, AB256126, AB256111, AB256130, AB256140, AB256134, AB256131, AB25611, AB256108, AB256099, AB256086, AB256122, AB256093, AB256104, AB256074, AB256051, AB256061, AB256070, AB256057, AB25608, AB256090, AB256066, AB256504, AB256485, AB256478, AB256498, AB256511, AB256493, AB25645, AB256447, AB256422, AB256438, AB256414, AB256458, AB256453, AB256433, AB256411, AB256399, AB256381, AB256392, AB256406, AB256386, AB25637, AB256343, AB256367, AB256351, AB256324, AB256358, AB256375, AB256332, AB256311, AB256316, AB256296, AB256292, AB256305, AB256263, AB256283, AB256270, AB256276, AB256255, AB256258, AB256242, AB256237, AB256236, AB256241, AB256252, AB256246, AB256231, AB256225, AB256215, AB256222, AB256228, AB256235, AB256204, AB256199, AB256194, AB256202, AB256207, AB256218, AB256176, AB256189, AB256181, AB256186, AB256192, AB256211, AB256173, AB256174, AB256170, AB256163, AB256159, AB256197, AB256097, AB256152, AB256050, AB256071, AB256111, AB256183, AB256091, AB256494, AB256434, AB256454, AB256414, AB256179, AB256474, AB256335, AB256393, AB256394, AB256374, AB256334, AB256275, AB256314, AB256294, AB256313, AB256333, AB256293, AB256273, AB256239, AB256253, AB256256, AB256214, AB256234, AB256174, AB256233, AB256194, AB256193, AB256213.
Para el HTLV-1 se incluyeron, en su orden, las secuencias cuyos códigos en el GenBank iban del AB114390 al AB114357 (Ozawa, et al., 2004), y del AY003899 al AY003886 (Cavrois, et al., 2000). Además, se seleccionaron las secuencias S80210, S80212 y S80213 (Cavrois, et al., 1995) y las AF228939 y DX597296 (Mortreux, et al., 2001). Se incluyeron, igualmente, 74 secuencias del genoma humano que flanquean repeticiones terminales largas (long terminal repeats, LTR), las cuales se obtuvieron mediante reacción en cadena de la polimerasa con transcripción inversa (RCP-TI) de sujetos seropositivos para el HTLV-1 provenientes de diferentes áreas de la costa pacífica colombiana (Cabrera, et al., 2004).
Alineamiento de las secuencias del genoma humano
Las secuencias de nucleótidos que flanquean las LTR de ambos tipos de provirus, se alinearon con aquellas del genoma humano (versión hg18) utilizando el programa BLASTn (http://www.ncbi.nlm.nih.gov/BLAST/). Con este procedimiento se encontraron 203 secuencias asignadas que reunieron los siguientes requisitos para ser consideradas como auténticos sitios de integración: (i) tener una secuencia de nucleótidos homóloga al terminal 3' LTR' del provirus del VIH-1 o el HTLV-1; (ii) tener al menos 95 % de identidad con secuencias del genoma humano versión hg18 que no presentasen discontinuidades; (iii) aparearse con una única secuencia del genoma humano cuyo porcentaje de identidad fuera el más alto registrado, y (iv) un tamaño mínimo de la secuencia seleccionada de 100 pares de bases.
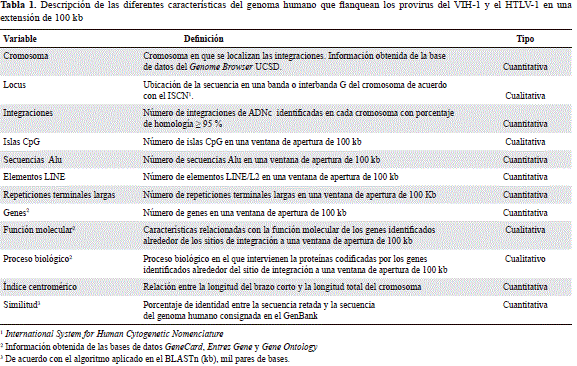
Análisis bioinformático de las secuencias
Para las secuencias seleccionadas la información sobre algunas características estructurales y funcionales de la cromatina asociada a la integración simultánea de ambos retrovirus se extrajo mediante las herramientas computacionales disponibles en el sitio de Internet de acceso libre del National Center for Biotechnology Information, NCBI, (http://www.ncbi.nlm.nih.gov) y del programa Genome Browser de la Universidad de California en Santa Cruz (http://genome.ucsc.edu/), también de acceso libre, en extensiones de los sitios de integración de 100 kpb. La información recolectada correspondió a la localización cromosómica, a los genes anotados (Refseq genes), a su función y proceso biológico, así como al contenido de islas CpG, y de secuencias Alu y SINE, entre otras (Tabla 1). Además, se obtuvo información de otras bases de datos como Gencard (versión 2.39) (http://www.genecards.org/cgi-bin/carddisp.pl), Gene Entrez (http://www.ncbi.nlm.nih.gov/ncbi/geneentrez) y Gene Ontology (GO) (http://www.geneontology.org/index.shtml).
Se utilizó la herramienta MEME suite (Multiple Em for Motif Elicitation) (http://nbcr-222.ucsd.edu/meme/) para obtener información de las secuencias motivo y las secuencias promotoras relacionadas con las integraciones provirales. La identificación de las secuencias promotoras en las que ocurrió la integración se obtuvo por similitud con las secuencias canónicas mediante el programa Tomtom Motif Comparison Tool (http://meme.ebi.edu.au/meme/tools/tomtom).
Distribución de la integración del VIH-1 y el HTLV-1 en el genoma humano
Para la distribución cromosómica de la integración de ambos retrovirus se tomó como referencia el patrón de bandas G y de interbandas propuesto por el Sistema Internacional para la Nomenclatura Citogenética Humana (International System for Human Cytogenetic Nomenclature, ISCN) (Simons, et al., 2013). A partir de esta información y empleando la correspondiente aplicación en el Genome Browser de la Universidad de California en Santa Cruz (http://genome.ucsc.edu/), se obtuvieron los datos correspondientes a la banda o interbanda G. A las zonas del genoma donde se registraron frecuencias elevadas de integración simultánea se les denominó zonas calientes de integración.
Del conjunto de datos consignados por She, et al. (2009) en el Gene Expression Omnibus de la plataforma, cuyo código de acceso GEO era GSE16546, se obtuvieron los datos de expresión génica de las muestras GSM413325 y GSM413332 para los linfocitos activados T CD4+ y T CD8+, respetivamente. Los datos de intensidad se utilizaron para calcular el valor del puntaje Z correspondiente a los genes analizados aplicando la siguiente expresión:
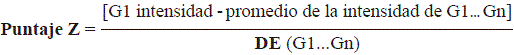
en donde G es el valor de la intensidad de fluorescencia de un gen dentro de la micromatriz, G1. . . Gn representa el valor promedio de la intensidad de todos los genes en la micromatriz de ADN y DE, la desviación estándar.
Además, para cada uno de los genes en los que ocurrieron integraciones simultáneas se obtuvieron datos sobre el tamaño y el número de exones y el tamaño del ARN mensajero a partir del Genome Browser.
Análisis estadísticos
Se hizo un análisis ortogonal de varianza de los factores principales utilizando el programa STATISTICA 7 (StataSoft Inc.NJ), para determinar las diferencias estadísticas en el patrón de integración simultánea de los dos retrovirus. Posteriormente se obtuvieron las diferencias en el número de provirus del VIH-1 y el HTLV-1 dentro de cada uno de los cromosomas mediante la prueba de diferencias significativas mínimas de Fisher, así como de las diferencias en su localización cromosómica y su ubicación telomérica o pericentromérica.
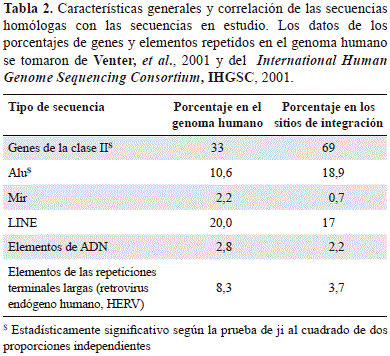
Las diferencias estadísticas de cada uno de los retrovirus con el tipo de secuencia adyacente a la LTR proviral (unidades de transcripción y las distintas familias de elementos repetidos), y su frecuencia en el genoma humano (International Human Genome Sequencing Consortium, 2001; Venter, et al., 2001), se calcularon con la prueba de ji al cuadrado, considerando como estadísticamente significativo un valor de p menor de 0,05. Posteriormente, se hizo una prueba exacta de Fisher con p menor de 0,05 para determinar las diferencias significativas con respecto a la función y los procesos biológicos asociados en aquellos genes localizados en las regiones genómicas en donde se determinó la integración del VIH-1 y el HTLV-1.
Resultados
Características de la integración del VIH-1 y el HTLV-1
De las 253 secuencias del genoma humano que flanquean las 3'LTR de los provirus del VIH-1 y el HTLV-1 escogidas, se obtuvieron 203 hits que coincidieron con regiones del genoma cuyas similitudes fueron ≥ 95 % y cumplieron con los requisitos establecidos previamente; 109 de ellas correspondieron al VIH-1 y 94 al HTLV-1. De este total de secuencias, el 47,3 % incluyó elementos repetidos del tipo Alu/LINE, SINE (short interspersed nuclear elements) y LTR, mientras que el 52,7 % restante correspondió a genes humanos de la clase II de referencia.
En todos los cromosomas, con excepción del Y, se registraron integraciones de ambos retrovirus. Sin embargo, el 46,61 % de los provirus del HTLV-1 se localizó en los cromosomas 1, 2, 5, 11 y 18, mientras que el 36,48 % de los del VIH-1 se integró en los cromosomas 2, 7, 12 y 17. Un análisis de los grupos evidenció patrones de integración diferentes para cada tipo de virus (prueba DMS = 5,77, gl: 46, p< 0,05) y entre cromosomas (ANOVA: p=0,04) (Figura 1).
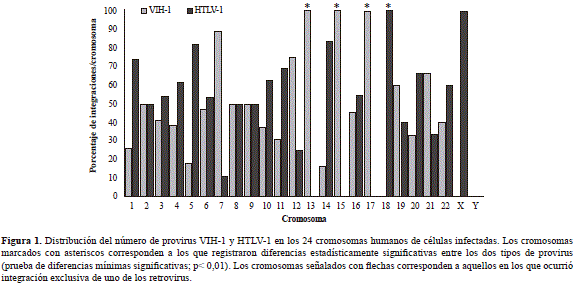
Se determinó la existencia de un proceso de exclusión cromosómica en la integración de ambos retrovirus en la muestra estudiada, lo que en el caso del HTLV-1 ocurrió en el cromosoma 18 (6 provirus), en tanto que en el caso del VIH-1 ocurrió en el cromosoma 17 (7 provirus) y en el 13 (1 provirus). Sin embargo, el 57,6 % (117/203) de las integraciones del VIH-1 y el HTLV-1 ocurrieron en los cromosomas 1 (10,90 %), 2 (10,32 %), 3 (6,10 %), 4 (6,10 %), 5 (5,63 %), 6 (7,05 %), 11 (6,10 %) y 12 (5,63 %), y en el resto fue variable. Los provirus del HTLV-1 se distribuyeron más frecuentemente en grupos, con una tendencia a la localización en regiones pericentroméricas (prueba de ji al cuadrado: gl:1; p<0,05), mientras que la distribución del VIH-1 se concentró preferencialmente en los telómeros (prueba de ji al cuadrado: gl:1; p<0,05).
Comparación de la distribución de la integración del VIH-1 y el HTLV-1 en el genoma humano
La clasificación de los elementos repetidos no codificantes asociados a los sitios de integración simultánea reveló que las secuencias de la familia SINE (73,8 %) y LINE (36,7 %) predominaban. En las SINE, las secuencias Alu correspondieron al 68,0 %, mientras que en las LINE, las de la familia L2 correspondieron al 7,3 %. El resto de elementos pertenecían a las LTR, los transposones ADN y las repeticiones simples (Figura 2).
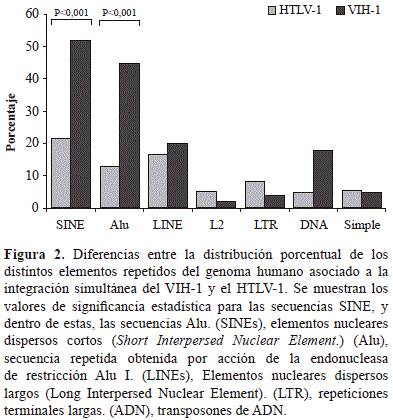
Con el objetivo de definir si existía una integración que no respondiera al azar en el genoma de los linfocitos humanos de ambos provirus, se aplicó la prueba de ji al cuadrado para analizar la distribución de la integración en los tipos de secuencias codificantes y no codificantes, así como en las repetidas obtenidas en este trabajo, y la de aquellas reportadas previamente para el total del genoma. En su conjunto, se observaron diferencias estadísticamente significativas entre la proporción del genoma humano que contiene ambos provirus y las secuencias codificantes (genes de clase II) y los elementos repetidos de la clase SINE, especialmente de la familia Alu, con el total del genoma (Tabla 2).
Distribución funcional de los genes adyacentes a los sitios de integración del VIH-1 y el HTLV-1
Del total de secuencias analizadas, el 52,7 % (107/203) correspondió a genes humanos clasificados en las diferen- tes categorías de Gene Ontology. Con respecto a su función molecular, no hubo diferencias estadísticamente significativas (prueba de ji al cuadrado: gl:3; p>0,05); las categorías funcionales más representativas fueron la unión a moléculas (52,4 %) y las enzimas que participan en varios procesos metabólicos (21,5 %) (Figura 3A).
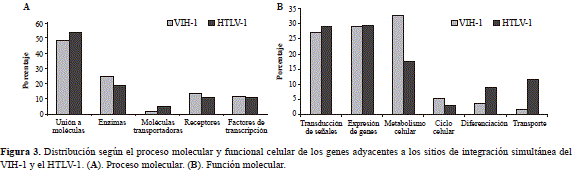
Con relación a los procesos moleculares, tampoco se encontraron diferencias significativas entre los dos virus en cuanto a la transducción de señales y la expresión de genes; sin embargo, en el caso de los genes asociados con el metabolismo se determinaron diferencias estadísticamente significativas entre ambos virus (prueba de ji al cuadrado: gl:1;p<0,05) (Figura 3B).
Características de la integración del HTLV-1 y el VIH-1 en zonas del genoma asociadas a los genes de clase II
El 56,9 % de las integraciones del VIH-1 ocurrió en genes de clase II codificantes para proteínas. Es interesante anotar que el 87,1 % del total de estas integraciones se registró en intrones (p<0,001). El 42,6 % de las integraciones del ADNc de HTLV-1 se detectó en genes clase II, y de estas el 67,5 % se ubicó en intrones (p<0,001). Se determinaron diferencias estadisticamente significativas en el número de intrones en los que ocurrió la integracion simultánea de los correspondientes ADNc virales. Se obtuvo una mayor frecuencia de integración de intrones en el VIH-1 que en el HTLV-1 (prueba de ji al cuadrado: p<0,05). Ningún provirus del VIH-1 se integró en elementos promotores del ARN polimerasa II, en contraste con los del HTLV-1, en el que el 20 % (8/40) correspondió a la integración en estas secuencias.
En general, se determinó que solamente se presentaron diferencias significativas en los tamaños de los genes asociados a las integraciones del VIH1 y el HTLV-1 (p<0,05), en contraste con el número de exones por gen y el tamaño de su correspondiente ARN mensajero.
Efecto en la expresión de los genes blanco de la integración del VIH-1 y el HTLV-1 en el ADN de diferentes poblaciones de linfocitos
Con el objetivo de analizar detalladamente las características de la integración simultánea del VIH-1 y el HTLV-1 en las distintas poblaciones de linfocitos, así como los efectos de la inactivación de los genes generados por la integración del ADNc de ambos retrovirus, se estudió la integración simultánea del VIH-1 y el HTLV-1 expresada conjuntamente en células T CD4+ y CD8+ normales. En el análisis de 40 genes asociados con la integración del HTLV-1, 16 (40,0 %) se expresaron en alguna de las dos poblaciones de linfocitos, en contraste con 29 (48,8 %) del VIH-1. Del total de genes en los que ocurrió la integración tanto del VIH-1 como del HTLV-1, el 15,7 % se expresó simultáneamente en algunas de las dos poblaciones de linfocitos no infectados por ambos retrovirus (Tabla 3).
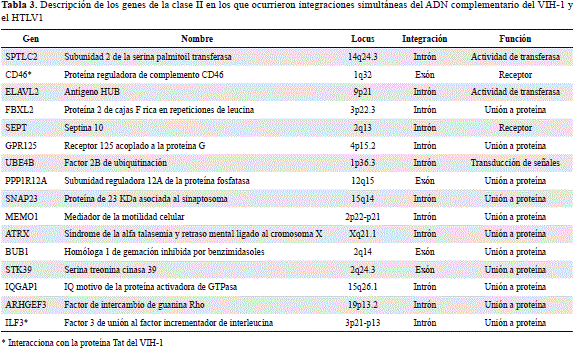
De acuerdo con el cálculo del puntaje Z, los genes CD46, SNAP23, ATRX, IQGAP1 e IFLR3 tuvieron los valores de expresión más elevados en las dos poblaciones linfocitarias analizadas (Figura 4). Es importante destacar que la integración del ADNc de ambos retrovirus en estos genes promueve la inactivación de su transcripción y produce efectos sobre la homeostasis de los linfocitos T4 y T8, afectando la expresión global de los genes en algunas de las rutas metabólicas comunes.
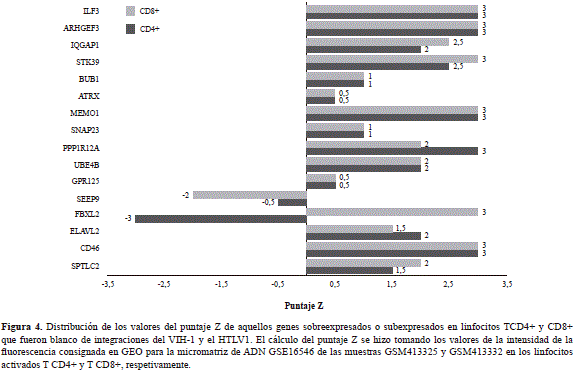
Entre los genes que presentaron el nivel de expresión más elevado estuvo el que codifica la proteína CD46. Esta molécula es un regulador del complemento que tiene funciones importantes relacionadas con la respuesta inmunológica. La proteína CD46 actúa como receptor de patógenos y es un potente estimulador de la respuesta de los linfocitos TH1 (Le Friec, et al., 2012). En las células infectadas por el VIH-1 la expresión del gen viral Nef promueve una baja regulación de la expresión de CD46 en la membrana plasmática, que afectaría la eficiencia de la infección (Haller, et al., 2014).
Discusión
La evidencia científica acumulada en los últimos 10 años indica que la integración proviral tanto del VIH-1 como del HTLV-I no se produce al azar, pues en dicho proceso las características estructurales y funcionales de la cromatina adyacente a los provirus en la célula hospedera podrían desempeñar un papel importante en la selección de los sitios de integración (Debyser, et al., 2015; Lewinski, et al., 2006; Hanai, et al., 2004; Tsukasaki, et al., 1997; Slattery, et al., 1999).
A partir de la simulación por computador efectuada en este trabajo, se obtuvo información que sustenta la hipótesis de que durante la infección simultánea por el VIH-1 y el HTLV-1 una combinación específica de características de la cromatina configuraría un ambiente genómico propicio para el proceso de integración de los dos ADNc retrovirales. Así, la densidad de islas CpG, de genes y de elementos repetidos, principalmente de secuencias Alu, revelaría las zonas del genoma humano en las que se facilitaría la integración del VIH-1 y el HTLV-1. De esta manera se confirman resultados previos de varios investigadores sobre las infecciones causadas por una sola especie (Weber, et al., 2014, Pavlice, et al., 2001; Craig & Bickmore, 1994).
En estudios anteriores se ha puesto de manifiesto la importancia de la estructura de la cromatina en la integración de los retrovirus (Wu & Burgess, 2004; Rynditcha, et al., 1998). Una conformación más relajada aumentaría la accesibilidad de los complejos de remodelación de la cromatina y del mismo complejo de preintegración proviral, variables que regularían la integración simultánea de los dos ADNc virales (Agbottah, et al., 2006). Una pregunta importante acerca de la integración del ADNc retroviral se refiere a si con los cambios topológicos de la cromatina, se genera un nuevo posicionamiento de loci. Los resultados de la integración del VIH-1 en diferentes genes humanos han demostrado un nuevo posicionamiento de los provirus en el interior del núcleo. Estos datos sustentan la propuesta de que la integración retroviral puede generar alteraciones en la organización de la cromatina y tener un efecto potencialmente modulador de la estructura cromatínica de la célula hospedera (Nagel, et al., 2012). Empleando la hibridación fluorescente in situ (FISH-3D), se ha demostrado que la integración proviral como factor epigenético podría estar implicada en la alteración de la función genómica (Gagniuc & Ionescu-Tirgoviste, 2013).
Los resultados estadísticos obtenidos para la muestra estudiada confirmaron las secuencias Alu como una de las variables que diseñarían el ambiente genómico preferido para la integración del ADNc retroviral. Varios de estos elementos han sido localizados en regiones teloméricas y sensibles al tratamiento con desoxirribonucleasa I, regiones que según nuestros resultados serían las preferidos para la integración del VIH-1 en contraposición con la tendencia del HTLV-1 a ubicarse en las zonas pericentroméricas. Estos hallazgos concuerdan con resultados previos en otros modelos de infección con una sola especie de retrovirus en los que se demuestra que las regiones blanco de la integra- ción son sensibles al tratamiento con esta enzima (Meekings, et al., 2008; Ciuffi, et al., 2006).
Las diferencias en los patrones de integración cromosómica de los dos retrovirus analizados estarían condicionadas por la dinámica de los telómeros durante el proceso de integración del VIH-1 y el HTLV-1. Aunque no hay información sobre el HTLV-1 en lo concerniente al efecto de la integración sobre los telómeros, en estudios anteriores se ha reportado que las poblaciones de linfocitos T de pacientes infectados por el VIH-1 sufren un acortamiento dinámico de sus telómeros, posiblemente debido a la expansión celular continua y prolongada que puede llevar a un agotamiento prematuro de la respuesta protectora antiviral y a una replicación senescente (Dagarag, et al., 2004; Effros, et al., 1996). Con base en los resultados obtenidos en este trabajo se podría sugerir que en las células del HTLV-1 infectadas por el VIH-1, el proceso de retrotransposición dependería de la disponibilidad de las zonas de cromatina internas debido a su tendencia a la concentración proviral no telomérica. Los telómeros son regiones dinámicas, compuestas por heterocromatina facultativa, que se activan o inactivan en fases definidas del desarrollo celular y que cambian activamente mediante la recombinación e integración de elementos transponibles (Riethman, 2008). Esta dinámica telomérica asociada a la integración diferencial observada condicionaría una integración del VIH-1 y el HTLV-1 estable en el tiempo (Nagel, et al., 2012; Gagniuc & Ionescu-Tirgoviste, 2013).
En este estudio los resultados del análisis de la simulación por computador coincidieron con aquellos reportados experimentalmente en infecciones causadas por una sola especie en lo relacionado con la selección del sitio blanco de la integración en regiones del genoma que poseen una elevada densidad génica de islas CpG y secuencias Alu (Derse, et al., 2007; Wang, et al., 2007; Barr, et al., 2006; Schroder, et al., 2002). Las islas CpG pueden someterse a metilación, lo que podría generar modificaciones epigenéticas locales (Hochstein, et al., 2007; Friedman, et al., 2011). Las secuencias Alu representan entre el 5 y el 10 % del genoma humano y juegan un papel crucial en los distintos eventos de reordenamiento cromosómico, así como en la regulación génica, pues se asocian con genes esenciales (housekeeping) y se localizan en regiones con alto contenido de islas CpG (Kumar, et al., 2007; Batzer & Deininger, 2002).
Además, las características funcionales de los genes localizados en la vecindad de los provirus del VIH-1 y el HTLV-1 coincidieron claramente con lo reportado en estudios previos en los que se plantea que los genes asociados con la transducción de señales y los procesos metabólicos celulares actuarían en la regulación de la expresión génica y en el transporte de moléculas como procesos fundamentales en el establecimiento de una integración estable del VIH-1 y el HTLV-1 (Eller, et al., 2007; Giri, et al., 2006).
Se determinó que los genes SNAP23, CD46, ATRX, IQGAP1, e ILF3R, ELAVL2, TUSC3, FBXL2, COL18A1, SEPT y GPR125 tuvieron una sobrexpresión conjunta en células TCD4+ yCD8+. Es importante destacar que la integración del VIH-1 y el HTLV-1 en los genes antes referidos generaría la supresión de su transcripción y, por lo tanto, la alteración de los perfiles de transcripción de las células con infección concomitante; ello conllevaría a una perturbación importante de la homeostasis, lo que se podría correlacionar con el efecto sistémico generado por la infección simultánea.
La integración del ADNc de los retrovirus en el genoma de la célula hospedera es de por sí un factor epigenético que promueve una remodelación de la cromatina asociada y también puede generar efectos a distancia. Con base en la información obtenida en este trabajo, es posible proponer un modelo en el que la integración del VIH-1 y el HTLV-1 estaría condicionada por aquellas regiones de cromatina interfásica localizadas en los territorios cromosómicos ricos en secuencias Alu, islas CpG y genes de clase II. Por efecto de la integración, se inducirían procesos de remodelación de la cromatina en zonas con elevada frecuencia de integraciones simultáneas del VIH1 y el HTLV1. Sin embargo, este no sería el único mecanismo que explicaría los patrones de distribución de la integración registrados en este trabajo; otros procesos, como los eventos epigenéticos locales ya descritos, tendrían efectos reguladores sobre aquellas zonas de la cromatina interfásica caracterizadas por una gran densidad de islas CpG y de genes (de Jong, et al., 2014). Sin embargo, y a pesar del volumen de evidencia científica que existe sobre este tema, faltan estudios que determinen cuál de todos los componentes que caracterizan los ambientes genómicos de la integración simultánea serían los principales efectores que influenciarían la dinámica total de este proceso.
Conflicto de intereses
Los autores declaramos que no existe ningún conflicto de intereses con respecto a los resultados de este trabajo.
Agradecimientos
El proyecto fue financiado por el Programa de Jóvenes Investigadores de Colciencias y como resultado de la convocatoria interna de la Vicerrectoría de Investigaciones de la Universidad del Valle (código Cicop.1728-2013).
Bibliografía
Ambrosi A., Cattoglio C., Di Serio C. 2008. Retroviral integration process in the human genome: Is it really non-random? A new statistical approach. PLoS Comput. Biol. 4: e1000144. [ Links ]
Barr S., Ciuffi A., Leipzig J., Shinn P., Ecker J., Bushman F. 2006. HIV Integration site selection: Targeting in macrophages and the effects of different routes of viral entry. Mol. Ther. 14: 218-25. [ Links ]
Batzer M.A. & Deininger P.L. 2002. Alu repeats and human genomic diversity. Nat. Rev. Genet. 3: 370-9. [ Links ]
Biasco L., Baricordi C., Aiuti A. 2012. Retroviral integrations in gene therapy trials. Mol. Ther. 20 (4): 709-16. [ Links ]
Brites C., Sampalo J., Oliveira A. 2009. HIV/human T-cell lymphotropic virus coinfection revisited: Impact on AIDS progression. AIDS Rev. 11 (1): 8-16. [ Links ]
Brites C., Harrington Jr. W., Pedroso C., Netto E.M., Badaró R. 1997. Epidemiological characteristics of HTLV-I and II coinfection in Brazilian subjects infected by VIH-1. Braz. J. Inf. Dis. 1: 42-7. [ Links ]
Bushman F., Lewinski M., Ciuffi A., Barr S., Leipzig J., Hannenhalli S., et al. 2005. Genome-wide analysis of retroviral DNA integration. Nat. Rev. Microbiol. 3: 848-58. [ Links ]
Cabrera-Moncayo J., Hiroshi M., Cerón F., Castillo A., Baba M., Akiba S., et al. 2004. Características moleculares de los sitios de integración del virus linfotrópico humano tipo I en linfocitos naturalmente infectados. Rev. Asoc. Col. Cienc. Biol. 16: 91-98. [ Links ]
Casoli C., Pilotti E., Bertazzoni U. 2007. Molecular and cellular interactions of VIH-1/HTLV coinfection and impact on AIDS progression. AIDS Rev. 9: 140-9. [ Links ]
Cattoglio C., Pellin D., Rizzi E., Maruggi G., Corti G., Miselli F., et al. 2010. High-definition mapping of retroviral integration sites identifies active regulatory elements in human multipotent hematopoietic progenitors. Blood. 116: 5507-17. [ Links ]
Cavrois M., Gessain A., Gout O., Wain-Hobson S., Wattel E. 2000. Common human T cell leukemia virus type 1 (HTLV-1) integration sites in cerebrospinal fluid and blood lymphocytes of patients with HTLV-1-associated myelopathy/tropical spastic paraparesis indicate that HTLV-1 crosses the blood-brain barrier via clonal HTLV-1-infected cells. J Infect Dis. 182: 1044-50. [ Links ]
Cavrois M., Wain-Hobson S., Wattel E. 1995. Stochastic events in the amplification of HTLV-I integration sites by linker- mediated PCR. Res Virol. 146: 179-8. [ Links ]
Ciuffi A., Mitchell R., Hoffmann C., Leipzig J., Shinn P., Ecker J., Bushman F. 2006. Integration site selection by HIV- based vectors in dividing and growth-arrested IMR-90 lung fibroblasts. Mol. Ther. 13: 366-73. [ Links ]
Craig J. & Bickmore W. 1994. The distribution of CpG in mammalian chromosome. Nature Genet. 7: 376-82. [ Links ]
Crise B., Y. Li C., Yuan D.R., Morcock D., Whitby D.J., Munroe L.O., et al. 2006. Simian immunodeficiency virus integration preference is similar to that of human immunodeficiency virus type 1. J. Virol. 79: 12199-204. [ Links ]
Dagarag M., Evazyan T., Rao N., Effros R.B. 2004. Genetic manipulation of telomerase in HIV-specific CD8+ T cells: Enhanced antiviral functions accompany the Iicreased proliferative potential and telomere length stabilization. J. Immunol. 173: 6303-11. [ Links ]
Daniel R. & Smith JA. 2008. Integration site selection by retroviral vectors: Molecular mechanism and clinical consequences. Hum. Gene Ther. 19: 557-68. [ Links ]
Debyser Z., Christ F., De Rijck J., Gijsbers R. 2015. Host factors for retroviral integration site selection. Trends Biochem. 40: 108-16 [ Links ]
Derse D., Crise B., Li Y., Princler G., Stewart C., Connor F., et al. 2007. HTLV-1 integration target sites in the human genome: Comparison with other retroviruses. J. Virol. 81: 6731-41. [ Links ]
Effros R.B., Allsopp R., Chiu C.P., Hausner M.A., Hirji K., Wang L., et al. 1996. Shortened telomeres in the expanded CD28- CD8- cell subset in HIV disease implicate replicative senescence in HIV pathogenesis. AIDS. 10: F17. [ Links ]
Eller C.D., Regelson M., Merriman B., Nelson S., Horvath S., Marahrens Y. 2007. Repetitive sequence environment distinguishes housekeeping genes. Gene. 390: 153-65. [ Links ]
Friedman J., Cho W.K., Chu C.K., Keedy K.S., Archin N.M., Margolis D.M., Karn J. 2011. Epigenetic silencing of VIH-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 85: 9078-89. [ Links ]
Gagniuc P & Ionescu-Tirgoviste C. 2013. Gene promoters show chromosome-specificity and reveal chromosome territories in humans. BMC Genomics. 14: 27. [ Links ]
Giri M.S., Nebozhyn M., Showe L., Montaner L.J. 2006. Microarray data on gene modulation by HIV-1 in immune cells: 2000-2006. J. Leukoc. Biol. 80: 1031-43. [ Links ]
Góngora-Bianchi R.A., Sosa-Cantón O., Pavía-Ruz N., Vera- Gamboa L., Lara-Perera D. 2003. Factores asociados con el riesgo de infección por retrovirus (VIH- 1 y HTLV-I/II) y su prevalencia en sexo trabajadoras de Campeche, México, en 1996-1997. Rev. Biomed. 14: 239-6. [ Links ]
Hacein-Bey-Abina S., Von Kalle C., Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., et al. 2003. LMO2- associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 302: 415-9. [ Links ]
Haller C., Müller B., Fritz J.V., Lamas-Murua M., Stolp B., Pujol F.M., et al. 2014. HIV-1 Nef and Vpu are functionally redundant broad-spectrum modulators of cell surface receptors, including tetraspanins. J Virol. 88: 14241-57. [ Links ]
Hanai S., Nitta T., Shoda M., Tanaka M., Isu N., Mizoguchi I., et al. 2004. Integration of human T-cell leukemia virus type I in genes of leukemia cells of patients with adult T-cell leukemia. Cancer Sci. 95: 306-10. [ Links ]
Hindmarsh P. & Leis J. 1999. Retroviral DNA integration. Microbiol. Mol. Biol. Rev. 63: 836-84. [ Links ]
Hochstein N., Muiznieks I., Mangel L., Brondke H., Doerfler W. 2007. The epigenetic status of an adenovirus transgenome upon long-term cultivation in hamster cells. J. Virol. 81: 5349-61. [ Links ]
Ikeda T., Shibata J., Yoshimura K., Koito A., Matsushita S. 2007. Recurrent HIV-1 integration at the BACH2 locus in resting CD4+ T cell populations during effective highly active antiretroviral therapy. J Infect Dis. 195: 716-25. [ Links ]
International Human Genome Sequencing Consortium. 2001. Initial sequencing and analysis of the human genome. Nature. 409: 860-921. [ Links ]
Kumar P.P., Mehta S., Purbey P.K., Ranveer D.N., Jayani S., Purohit H.J., et al. 2007. ATB1-binding sequences and Alu-like motifs define a unique chromatin context in the vicinity of human immunodeficiency virus type 1 Integration Sites. J. Virol. 81: 5617-27. [ Links ]
Laurentino R.V., Lopes I.G., Azevedo V.N., Machado L.F., Moreira M.R., Lobato L., et al. 2005. Molecular characterization of human T-cell lymphotropic virus coinfecting human immunodeficiency virus 1 infected patients in the Amazon region of Brazil. Mem. Inst. Oswaldo Cruz. 100: 371-6. [ Links ]
Lefrère J.J., Couroucê A.M., Mariotti M., Wattel E., Prou O., Bouchardeau F., et al.1990. Rapid progression to AIDS in dual VIH-1/HTLV-I infection. Lancet. 336: 509. [ Links ]
Lewinski M.K., Yamashita M., Emerman M., Ciuffi A., Marshall H., Crawford G., et al. 2006. Retroviral DNA integration: Viral and cellular determinants of target-site selection. PLoS Pathogens. 2: e60. [ Links ]
Le Friec G., Sheppard D., Whiteman P., Karsten C.M., Shamoun S.A., Laing A., et al. 2012. The CD46-Jagged1 interaction is critical for human TH1 immunity. Nat Immunol.13: 1213-21. [ Links ]
Meekings K.N., Leipzig J., Bushman F.D., Taylor G.P., Bangham C. 2008. HTLV-1 integration into transcriptionally active genomic regions is associated with proviral expression and with HAM/TSP. PLoS Pathol. 4: e1000027. [ Links ]
Mitchell R.S., Beitzel B.F., Schroder A.R., Shinn P., Chen H., Berry C.C., et al. 2004. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2: e234. [ Links ]
Moriuchi H., Moriuchi M., Fauci A.S. 1998. Factors secreted by human T lymphotropic virus type I (HTLV-I)-infected cells can enhance or inhibit replication of VIH-1 in HTLV- I-uninfected cells: Implications for in vivo coinfection with HTLV-I and VIH-1. J. Exp. Med. 187: 1689-97. [ Links ]
Mortreux F., Leclercq I., Gabet A.S., Leroy A., Westhof E., Gessain A., et al. 2001. Somatic mutation in human T-cell leukemia virus type 1 provirus and flanking cellular sequences during clonal expansion in vivo. J Natl Cancer Inst. 93: 367-77. [ Links ]
Nagel J., Gross B., Meggendorfer M., Preiss C., Grez M., Brack-Werner R., et al. 2012. Stably integrated and expressed retroviral sequences can influence nuclear location and chromatin condensation of the integration locus. Chromosoma. 121 (4): 353-67. [ Links ]
Nienhuis A.W., Dunbar C.E., Sorrentino B.P. 2006. Genotoxicity of retroviral integration in hematopoietic cells. Mol. Ther. 13: 1031-49. [ Links ]
Ozawa T., Itoyama T., Sadamori N., Yamada Y., Hata T., Tomonaga M., et al. 2004. Rapid isolation of viral integration site reveals frequent integration of HTLV-1 into expressed loci. J Hum Genet. 49: 154-65. [ Links ]
Pavlice K.A., Jabbari K., Paces J., Hejnar J., Bernardi G. 2001. Similar integration but different stability of Alus and LINEs in the human genome. Gene. 276: 39-45. [ Links ]
Pedroso C., Netto E.M., Weyll N., Brites C. 2011. Coinfection by VIH-1 and human lymphotropic virus type 1 in Brazilian children is strongly associated with a shorter survival time. J. Acquir. Immune Defic. Syndr. 57 Suppl 3: S208-11. [ Links ]
Pilotti E., Bianchi M.V., De Maria A., Bozzano F., Romanelli M.G., Bertazzoni U., et al. 2013. HTLV-1/-2 and VIH-1 co-infections: Retroviral interference on host immune status. Front. Microbiol. 4: 372. [ Links ]
Pryciak P.M. & Varmus H.E. 1992. Nucleosomes, DNA-binding proteins, and DNA sequence modulate retroviral integration target site selection. Cell. 69: 769-80. [ Links ]
Riethman H. 2008. Human telomere structure and biology. Annu, Rev. Genomics Human Genetics. 9: 1-19. [ Links ]
Rynditcha A., Zoubaka S., Tsybaa L., Tryapitsina-Guleya N., Bernardi G. 1998. The regional integration of retroviral sequences into the mosaic genomes of mammals. Gene. 22: 1-16. [ Links ]
Salcedo-Cifuentes M., Domínguez M.C., García-Vallejo F. 2011. Genomic epidemiology of the HTLV-1 integration process in TSP/HAM cases. Pan Am. J. Public Health. 30: 422-30. [ Links ]
Schroder A.R., Shinn P., Chen H., Berry C., Ecker J.R., Bushman F. 2002. VIH-1 integration in the human genome favors active genes and local hotspots. Cell. 110: 521-29. [ Links ]
She X., Rohl C.A., Castle J.C., Kulkarni A.V., Johnson J.M., Chen R. 2009. Definition, conservation and epigenetics of housekeeping and tissue-enriched genes. BMC Genomics. 10: 269. [ Links ]
Sierra S., Kupfer B., Kaiser R. 2005. Basics of the virology of VIH-1 and its replication. J. Clin. Virol. 34: 233-44. [ Links ]
Simons A., Shaffer L.G., Hastings R.J. 2013. Cytogenetic Nomenclature: Changes in the ISCN 2013 Compared to the 2009 Edition. Cytogenet. Genome Res. 141: 1-6. [ Links ]
Slattery J., Franchini G., Gessain A. 1999. Genomic evolution, patterns of global dissemination, and interspecies transmission of human and simian T-cell Leukemia/ Lymphotropic Viruses. Genome Res. 9: 525-40. [ Links ]
Soto J., Peña A., García-Vallejo F. 2011. A genomic and bioinformatics analysis of the integration of HIV in peripheral blood mononuclear cells. AIDS Res. Hum. Retroviruses. 27: 547-55. [ Links ]
Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D., et al. 2004. A gene atlas of the mouse and human protein- encoding transcriptomes. Proc. Natl. Acad. Sci. USA. 101: 6062-7. [ Links ]
Tsukasaki K., Tssima H., Yamamura M., Hata T., Murata K., Maeda T., et al. 1997. Integration patterns of HTLV-I provirus in relation to the clinical course of ATL: Frequent clonal change at crisis from indolent disease. Blood. 89: 948-56. [ Links ]
Van Maele B., Busschots K., Vandekerckhove L., Christ F., Debyser Z. 2006. Cellular co-factors of VIH-1 integration. Trends Biochem. Sci. 31: 98-105. [ Links ]
Venter J.C., Adams M.D., Myers E.W., Li P.W., Mural R.J., Sutton G.G., et al. 2001. The sequence of the human genome. Science. 291: 1304-51. [ Links ]
Wang G.P., Ciuffi A., Leipzig J., Berry C.C., Bushman F.D. 2007. HIV integration site selection: Analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Res. 17: 1186-94. [ Links ]
Weber S., Weiser B., Kemal K.S., Burger H., Ramírez C.M., Korn K., et al. 2014. Epigenetic analysis of VIH-1 proviral genomes from infected individuals: predominance of unmethylated CpG's. Virology. 449: 181-9 [ Links ]
Wu X. & Burgess S.M. 2004. Integration target sites selection for retroviruses and transposable elements. Cell. Mol. Life Sci. 61: 2588-96. [ Links ]
