










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Academia Colombiana de Ciencias Exactas, Físicas y Naturales
Print version ISSN 0370-3908
Rev. acad. colomb. cienc. exact. fis. nat. vol.40 no.156 Bogotá July/Sept. 2016
https://doi.org/10.18257/raccefyn.364
doi: http://dx.doi.org/10.18257/raccefyn.364
Laboratory of Molecular Biology and Pathogenesis, Universidad of Valle, Cali, Colombia. Corresponding author: Felipe García-Vallejo, labiomol@gmail.com.
Inaugural article by number member of the Colombian Academy of Exact, Physical and Natural Sciences in May 13, 2016.
Received: April 13, 2016. Accepted: August 10, 2016
Abstract
Introduction. The distribution of human lentiviral cDNA into the host genome has been studied using a linear structural approach, however such analysis is incomplete because do not consider the dynamics and topology of interphase chromatin and the gene expression networks in infected cells.
Objective. To correlate using a non-linear approach the multifractality of human chromosomes, with the composition and disturbing of chromatin topology, as complex effect promote by the lentiviral cDNA integration.
Methods. From 2,409 human genome sequences flanking the 5'LTR of human and simian lentiviruses obtained from GeneBank (NCBI) database, several human genomic variables were correlated with the multifractality values AvDDq of chromosomes covering more than 98.6% of the human genome. Moreover Cytoscape v.2.63 was used to simulate the effects of viral cDNA integration on gene expression networks in macrophages.
Results. The 54.21% of lentivirus cDNA integrations were registered in chromosomes with high and medium fractality; 18.14% of these cDNA integrations was exclusively located in chromosomes 16, 17, 19 and 22 corresponding to that with high multifractality values. High scores of Pearson's correlation for AvDDq/ chromosome vs integrations/chromosome; percentage of Alu sequences were recorded. 2,770 interactions among 28 genes located closed to HIV-1 proviruses in human macrophages were recorded. cDNA integration alters the gene interaction networks in infected cells, the topological parameters of non-infected macrophage network gene was dramatically changed upon HIV-1 integration.
Conclusion. Some topological changes in those regions with high frequency of cDNA viral integrations would synergistically configure local topological chromatin environments that alters the gene interaction networks in infected cells.
Key words: Retrovirus; Retroviral co-integration; Lymphocytes; CpG Island; Class II Genes; Computational simulation.
Resumen
Introducción. La distribución del DNAc lentiviral en el genoma hospedero ha sido estudiada usando un enfoque estructural, sin embargo éste es incompleto pues no considera la dinámica y la topología de la cromatina interfásica y las redes de expresión de genes en la célula infectada.
Objetivo. Utilizando un enfoque no linear, correlacionar la multifractalidad de los cromosomas humanos con la composición y el disturbio de la topología de la cromatina como un efecto complejo promovido por la integración de ADNc lentiviral.
Métodos. De 2.409 secuencias genómicas obtenidas del GeneBank y flanqueantes al 5'LTR de lentivirus humanos, (cobertura mayor del 98,6% del genoma humano), se correlacionaron con los valores de multifractalidad (AvDDq) de la cromatina humana. Adicionalmente se empleó el programa Cytoscape v.2.63 para simular computacionalmente los efectos de la integración sobre las redes de expresión de genes humanos.
Resultados. El 54,21% de la integración lentiviral ocurrió en aquellos cromosomas con valores altos e intermedios de multifractalidad; el 18.14% de estas integraciones se localizo en los cromosomas con más altos valores de multifractalidad (16, 17, 19, 22). La multifractalidad se correlacionó con el porcentaje Alu. Se registraron 2.770 interacciones entre 28 genes localizados cerca de provirus VIH-1 en macrófagos humanos. La integración del DNAc lentiviral alteró dramáticamente, la topología de la red de expresión de genes en macrófagos.
Conclusión. Algunos cambios topológicos asociados a las regiones con elevada frecuencia de integración del ADNc, podrían, de manera sinérgica, reconfigurar localmente la topología del ambiente cromatínica que las redes de expresión de genes en la célula infectada.
Palabras clave: Retrovirus; Cointegración retroviral; Linfocitos; Isla CpG; Genes clase II; Simulación computacional.
Introduction
The study of particular systems is the source of inspiration that guides the formation of general ideas from specific cases to general principles. Therefore the systemic approach extends towards the study of fundamental biological questions, such as gene assembly, protein folding and the nature of functional specificity (Diehl y Boyle, 2016). Such issues extend beyond the current perception of bioinformatics as a support discipline and address aspects of biological complexity, including the simulation of molecular interaction networks and the dynamics of chromatin using nonlinear methods (Polychronopoulos, et al, 2016; Karimi-Busheri y Rasouli-Nia, 2015).
The two closely related human lentiviruses HIV-1 and HIV-2 are responsible for the 21th century AIDS pandemic (Piot, et al, 2015; UNAIDS, 2013). Most current therapeutic approaches use combinations of antiviral drugs that inhibit activities of viral enzymes such as reverse transcriptase, protease and integrase; nevertheless none of those have succeeded in controlling infection (Margolis, et al, 2015; Balakrishnan, 2009; Wang, et al, 2010; Zaccarelli, et al, 2005). One option to overcome the problem is to explore new therapies that include the study of the viral cDNA integration dynamics of human Lentiviruses. It would permit to understand the underpinnings behinds of alterations of cellular homeostasis when a cell is infected (Moore, 2000). Additionally, analysis of integration process is important in HIV-induced disease, in Lentivirus-based gene therapy and new therapeutic options actually developed (Saayman, et al, 2015; Cereseto y Giacca, 2004).
Integration is a crucial step in the life cycle of retrovirus permitting the incorporation of viral cDNA into the host genome (Coffin, 1996; Hindmarsh y Leis, 1999). Lenti- viral cDNA integration is mediated by the virally encoded integrase enzyme and other viral and cellular proteins in a molecular complex called the pre-integration complex (PIC) (Le Sage, et al, 2014; Van Maele, et al, 2006). Previously Lentiviral integration process has been extensively studied using a wide array of molecular biology, biochemistry and structural biology approaches [Lewinski, et al, 2006]. However, is critical to directly identify the viral distribution inside human genome in order to understand, at genomic, level the relationships between the composition and topology of chromatin with the target sites selection (Wong, et al, 2015; Soto-Girón and García-Vallejo, 2012).
As shown by previous studies, the preferences in target site selection for integration are not entirely random (Ferris, et al, 2010; Jordan, et al, 2001; Wu, et al, 2003; Hematti, et al, 2004; Soto, et al, 2011); being pronounced favored and disfavored chromosomal regions which differ among retroviruses (Benleulmi, et al, 2015; Rick, et al, 2004). These preferential regions of host genomes are characterized by having a high frequency of integrational events, as known as âhotspotâ and are distributed along the genome of host cell (Maxfield, et al, 2005; Soto, et al, 2010). In HIV-1, most of proviruses are localized into transcriptionally active regions not only in exons and introns, but also in sequences around start transcription sites (Schroder, et al, 2002; Derse, et al, 2007).
As the scenario of retrovirus cDNA integration process is the human genome at once, it is necessary to consider it as one of a complex molecular structures ever seen in nature. Its extraordinary information content has revealed a surprising mosaicims between coding and non-coding sequences (Moraes and Góes, 2016; Lander, et al, 2001; Venter, et al, 2001; Levy, et al, 2007). This highly regionalized structure introduces complex patterns for understanding the gene structure and repetitive DNA sequence composition providing a new scenario to study biological process such as Lentivirus cDNA integration and its consequences into host genome.
The human genome coding regions are defined, in part, by an alternative series of motifs responsible for a variety of functions that take place on the DNA and RNA sequences, such as, gene regulation, RNA transcription, RNA splicing, and DNA methylation. The sequencing of the human genome revealed a controversial number of interrupted genes (approximately 26,000) with their regulatory sequences [Venter, et al, 2001] representing about 2% of the genome. These genes are immersed in a giant sea of different types of non-coding sequences which make up around 98% of the genome. The non-coding regions are characterized by many kinds of repetitive DNA sequences, where almost 10.6% of the human genome consists of Alu sequences, a type of SINE (short interspersed elements) sequence (ENCODE Project Consortium, 2007; Versteeg, et al, 2003). They can act as insertional mutagens and the vast majority appears to be genetically inert (DeCerbo y Carmichael, 2005). LINES, MIR, MER, LTRs, DNA transposons, and introns are other kinds of noncoding sequences, which together conform about 86% of the genome. In addition, some of these sequences are overlapped one to another, for example, the CpG islands (CGI), which complicates analysis of the genomic landscape. In turn, each chromosome is characterized by some particular properties of structure and function (The ENCODE Project Consortium, 2012; ENCODE Project Consortium, 2007).
Beyond the linear approach of the retroviral cDNA integration process, is also necessary to understanding in toto all variables involved in the process. To extend the knowledge about the complexity of integration process, a new field of analysis by using nonlinear methods is necessary. One of such approach is to apply the fractal analysis, which put in context the variable dynamics of chromatin topology as a new scenario to better understand the process of lentiviral integration (Yi, et al, 2015). In this sense our group have previously reported the fractality in the human genome sequence. This behavior correlates strongly on the presence of Alu elements and to a lesser extent on CpG islands and (G+C) content. In contrast, no or low relationship was found for LINE, MIR, MER, LTRs elements and DNA regions poor in genetic information. Gene function, cluster of orthologous genes, metabolic pathways, and exons tended to increase their frequencies with ranges of multifractality and large gene families were located in genomic regions with varied multifractality (Moreno, et al, 2011). Furthermore, the multifractal analysis of the human genome permitted to classify the human chromosomes into three different groups according with their multifractality: The first group were chromosomes 19, 17, 22 and 16 that exhibit the highest multifractality values and also the highest Alu contents; a second group included chromosomes 15, 20, 1, 10, 12, 9, 7, 14 and 21 which have medium multifractality; and a third group of chromosomes 2, 11, 8, 6, Y, 3, 18, 5, 13, X, and 4 with the lowest multifractality (Moreno, et al, 2011).
In the current work I report the complexity of the human cDNA lentivirus integration process using different sources of genome information obtained from several genome data-bases. The computational simulation in association with the use of strong statistical methods and a chaos game algorithm, revealed the high level of complexity of lentiviral cDNA integration which is dependent on not only of the genome structure itself but the dynamics and topology of host human chromatin. As a general conclusion of the analyses was the close association between high frequency of lentivirus integration zones in that human chromosomes with high multifractality as a potential effect of chromatin regions that are far from of equilibrium.
Methods
Mapping of Lentivirus integration sites in the human genome.
A total of 2,409 human genome sequences flanking the 5'LTR of human and simian lentiviruses were obtained from GenBank (NCBI) database. They were previously deposited by several authors using different experimental approaches and also several type of host cells (table 1) (Ikeda, et al, 2007; Crise, et al, 2005; Mitchell, et al, 2004; Wu, et al, 2003; Schröder, et al, 2002). Using the BLASTN algorithm (http://blast.ncbi.nlm.nih.gov/Blast.cgi), all sequences were aligned to the draft human genome (hg18) and those that met the following criteria were considered authentic integration sites: (i) contained the terminal 3' end of the HIV-1 HIV-2, SIV or HIV vectors LTR. (ii) had matching genomic DNA within five base pairs of the end of the viral LTR. (iii) had at least 95% homology to human genomic sequence across the entire sequenced region. (iv) matched a single human genetic locus with at least 95% homology across the entire sequenced region. (v) had minimum size of 150 bp. Initially the chromosomal band and the location of the sequence under analysis were obtained from de NCBI map viewer (http://www.ncbi.nlm.nih.gov/mapview/).
Several genomic variables were counted from different files: C+G and CpG islands from seq_cpg_islands.gz file, Alu, LINEs from seq_repeat.md.gz file, genes and genes/Mbp from seq_gene.md.gz file. All these files were downloaded from NCBI human build 36.2. The chromosomal localization of the lentiviral provirus included in the present study was performed by using the G pattern banding of each chromosome according with the International System for Human Cytogenetic Nomenclature -ISCN- (Simonis, et al, 2006). The mean values of multifractality (AvDDq) per each chromosome were obtained from the analysis of the multifractality that included 9,389 genomic fragments of 300 Kb each one, representing 2,816,700 Kb covering more than 98.6% of the human genome (Moreno, et al, 2011). Protein network construction
To construct a gene expression network associated with HIV-1 integration process, we selected 28 genes of human monocytes/macrophages, located in the vicinity of HIV-1 integration sites and previously reported by (Soto, et al, 2010): AXIN1, NFAT5, STAT5A, FLT1, AKT3, HTT, RIPK2, DGCR8, WWOX, NRG1, DYRK1A, SLC2A14, STAP1, ZNF36, PKD1, STX1A, RACGAP1, ATF-7, EXT1, NOLC1, TCF4, PSMD13, RBMS3, CENTD1, ACAA2, NFAT5, PI4KA, FLT1.
Cytoscape v.2.63 (Shannon, et al, 2003) was used to construct a gene expression network from two kinds of files: The first one from gene expression profiles as a text file (.pvals) that were imported of expression data microarray experiments (GEO profiles, NCBI). The second, as data annotation in text files (.sif) that corresponds to each one gene-gene interactions (online databases). In the first one, gene expression values were collected from the microarray data series GSE19236 composed by two Agilent platforms (GPL6480 and GPL6848) with 48 samples of monocytes to macrophages, macrophages and dendritic cells. These data were previously reported (Klug, et al, 2010). Samples from healthy donors were taken and analyzed using GeneSpring GX 10.0.2 (Agilent). Data were normalized to the 75th percentile and baseline-transformed to the median of freshly isolated monocyte samples. These are available from the National Center for Biotechnology Information (NCBI), Gene Expression Omnibus (GEO) repository (accession number GEO: GSE19236). For network analysis, all macrophages expression samples with accession code GSM476720, GSM476721, GSM476722, GSM476723, GSM476724, and GSM476725 were selected.
To make the corresponding statistical analyses Multi Experiment Viewer v4.1 was applied (Saeed, et al, 2006). Using data from Biomolecular Network Data Bank (BOND, http://bond.unleashedinformatics.com/Action), Biological General Repository for Interaction Datasets (BioGird, http://thebiogrid.org/) and Kyoto Encyclopedia of Genes and Genomes (KEEG, http://www.genome.jp/kegg/), a new file with the interaction data of 28 genes located close to integration sites was constructed.
Functional analysis
Cytoscape v2.6 was used for visualizing and analyzing the genetic interaction networks among 28 human monocyte/ macrophages genes and their interactions. Biological Networks Gene Ontology v2.6 plugin (BiNGO tool) was used to determine which gene ontology (GO) terms are significantly overrepresented in a set of genes. A hypergeometric test was applied to determine which categories were significantly represented (p-value <0.01); significant value was adjusted for multiple hypotheses testing using the Bonferroni family wise error rate correction (Maere, et al, 2005).
The network topology parameters were calculated using network analyzer plugin, which includes network diameter, the number of connected pairs of nodes and average number of neighbors; it also analyses node degrees, shortest paths, clustering coefficients, and topological coefficients (Max Planck Institute Informatik).
To identify active sub-networks as highly connected regions of the main network we used jActiveModules plug- in that grouped genes according with significant p-values of gene expression over particular subsets of samples. The result shows active modules, listed according to the number of nodes, and an associated Z-score. An active module with Z-scores greater than 3.0 indicated significant response upon the conditions of the experiment. We kept the standard default values, as being the most effective for initial analyses (Ideker, et al, 2002).
To compare the currently networks (normal and simulated) to a randomized versions of itself, I performed a comparison analysis to random network analysis using the Random Network v.1.0 plugin of cytoscape previously described by Mcsweeney, (2008). This plugin creates a number of randomized networks and analyzing several metrics parameters. The metric parameters that we used were clustering coefficient (measures how close the neigh-borhood of a node is to being complete) and the average degree (number of edges divided by number of nodes) and the threads to run was 2 and the rounds to run (number of randomizations to perform) were 100.
Effect of cDNA integration on the monocyte macrophage network
One purpose of the study was to simulate the effect of inactivation of some sets of genes that were previously reported as target for proviral integration in non-infected macrophages (Barr, et al, 2006).The effect of gene silencing was made by turning off 5 genes with the highest number of interactions (AXIN1, NFAT5, AKT3, FLT1, and STAT5A), to remove them from the data matrix, that operation simulate a HIV-1 infected network in monocyte/macrophage.
Statistical analyses
Bivariate and multivariate analysis were carried out using STATISTICA 7.30. Linear correlation analyses for chromosomal integration vs multifractality per chromosome (AvDDq) were performed; chromosomal integration vs Alu, CpG island distribution and gene content per chromosome were calculated; additional a non-parametric regression was calculated for AvDDqvs total lentivirus integration per chromosome. For the multivariate analyses, we included 10 genomic variables which are described in table 2. The fitness of variables in the Principal Component Analysis (PCA) model was evaluated by the calculus of the Bartlett's sphericity test as well as by Eigen values; the number of components included in the study was determined considering a value of variance over 75%. The significant statistical correlation among the genomic variables including the AvDDq was calculated by a Pearson's correlation matrix. A p-value <0.05 was considered as significant for all tests performed.
To identify which genes were significant among samples in microarrays an ANOVA test was calculated, considering a p-value <0.001 as significant. Additionally, a hierarchical clustering analysis of the samples using Euclidean Distance Method and mean linking were performed.
Statistical differences in the network's topological parameters (clustering coefficient, shortest paths, network heterogeneity, the centralization, average number of neigh-bors and characteristic path lengt), was performed using Statistic 7 (STATSOFT, INC., 2004) applying a Kolmogorov-Smirnov test, with a significance value at p<0.05.
Results
Patterns of provirus distribution
No gross significant differences were observed in the integration lymphocytic profiles between HIV-1 and HIV-2 (p>0.05, Mann-Whitney test). The integrational events for both human Lentiviruses were recorded in all chromosomes except the Y (figure 1). However, significant differences between the number of HIV-1 and HIV-2 provirus were observed for chromosomes 4, 8, 9, 11 and 16 (p<0.05, X2 test). The 12% (289/2,409), of total integrations occurred in chromosome 17 (figure 1). Moreover a tendency to a differential distribution of provirus towards telomeric and subtelomeric regions of the most of human chromosomes was observed. In this sense, other authors showed that cen- tromeric alphoid repeat regions are disfavored as integration sites (Carteau, et al, 1998). Although proviruses were observed in all chromosomes, we identified some chromatin regions with only HIV-1 integrations in chromosomes 4, 6 and 9 and only HIV-2 in chromosome 21.
Definition of the common genomic microenvironments for integration
The results of multiple regression analysis conducted on the HIV-1 and HIV-2 data sets showed that there were differential distributions of CpG island, genes, and Alu elements that together conditioned a specific genomic environment per chromosome (R2=0.91, p<0.05). Gene density was the independent variable contributed most in the prediction of the dependent variable (integrations) due to the highest regression coefficients (B=0.83; p<0.05). The highest relative likelihood of hosting a lentiviral integration event in the human genome was registered in chromosome 17 (figure 2a).
To test weather integration events are favored by gene-rich regions in all chromosomes, a comparison between those variables was done indicating that a high gene density in chromatin regions determine a favorable environment for integration, even when the chromosome 17 is excluded (figure 2b). Because chromosome 17 registered the highest percentage of lentiviral integration events, a detailed analysis of chromatin structure correlating several variables that give data about the cellular chromatin status was performed. In general the distal chromatin regions of p and q arms showed similarities in the distribution of methylation in CpG islands, methylation in several lysine residues of histone H3 (K4, K27 and K36) and variable levels of open chromatin and nucleosome occupancy.
cDNA integration and chromosomal multifractality
The 54.21% (1,306/2,409) of lentivirus cDNA integrations were registered in chromosomes with high and medium fractality; however 18.14% (437/2,409) of these cDNA integrations was located in chromosomes 16, 17, 19 and 22 corresponding to that with high multifractality values (figure 3). High scores of correlation coefficients for number of integrations/chromosome vs MIM diseases (r=0.773); CpG islands (r=0.888) and genes (r=0.895); AvDDq/chromosome vs MRTR (0.938); integrations/chromosome (0.620); per-centage of Alu sequences (0.97) and genes/Mb (0.873) were recorded (table 3). Furthermore in chromosome 17 with an AvDDq=1.1572 and a gene density of 17.88 genes/Mb, had the highest frequency of lentiviral cDNA integrations.
Although the association between multifractality per chromosome and cDNA integration profiles were almost similar for HIV-1, HIV-2, HIV vector and SIV, some minor differences were recorded. The high frequencies of integration in chromosomes 17 and 19 were similar in HIV-2, HIV-vector and SIV; however, in chromosomes with medium fractality especially in 1 and 7, integration frequencies were different for HIV-1 and HIV-2 in com-parison with HIV-vector and SIV (figure 4).
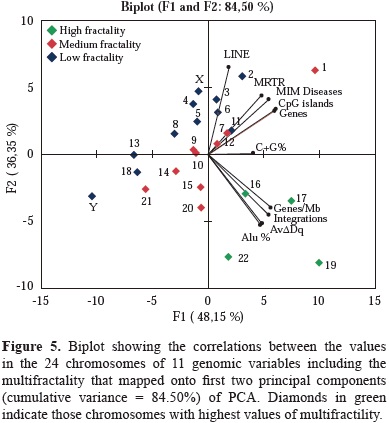
The results of the Bartlett's sphericity test (p<0.05) and of those obtained in the correlations matrix test, showed a high correlation among the selected genome characteristics under analysis (table 2). The data indicated that the most appropriate multivariate analysis for the hierarchical statistical processing of variables was PCA. It rendered two combinations of genomic variables that accounted for the 84.50% of the total variance. The first component summarized 48.15% of the total variance, and the second 36.35%. The first one was a linear combination of variable numbers per chromosome, of MIM diseases, cDNA integrations, AvDDq, Alu, CpG and Genes per Mb, while the second component included, number of LINE and genes per chromosome. As shown in figure 5, AvDDq, Alu %, Genes/Mb and integrations co-localized in the same Cartesian quarter with high multifractal chromosomes 16, 17, 19 and 22.
The effect of cDNA integration in monocyte/macrophage protein networks
11,713 significant genes of 41,000 probes were clustered in two significant different groups of cells; one of them included only dendritic cells, meanwhile the second grouped monocyte to macrophages and macrophages which are sharing similar gene expression patterns. A total of 2,770 interactions among 28 genes which were located closed to HIV-1 proviruses in human macrophages were recorded. AKT3 was gene with highest number of interactions (456), followed by FLT1 (381), STAT5A (356) and AXIN1 (328). In contrast ZNF36, DYRK1A and RBMS3 genes had the lowest number of gene interactions. The normal monocyte/ macrophage gene network showed tree components: the main cluster composed by 26 genes and its interactions and two minor clusters in which ZNF36 gene was the central node with five interactions; and STX1A as central node with twelve interactions.
Using the KEGG Pathway Database, 38 pathways were associated to those 28 macrophages genes. This metabolic signature contained pathways closely articulated with Wnt, Jak-STAT, ErbB, and VEGF signaling pathways. The gene transcriptional products involved in these cascades leads to different signaling pathways, resulting in multitude of signals for cell proliferation, differentiation and cellular homeostasis.
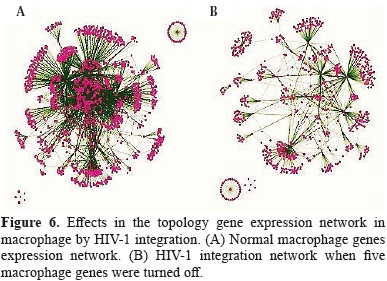
In order to better understand the alteration of monocyte/macrophages homeostasis by the HIV-1 integration, our analyses were focused to simulate what are the effects of viral cDNA integration in the alteration of several gene expression networks in human macrophage. In general the topology of non-infected macrophage network gene was dramatically changed by the HIV-1 integration events that lead to turned off the expression of five genes by the integration of proviral cDNA.
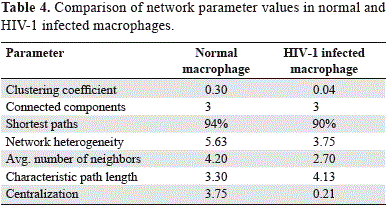
The evaluation of the several topological parameters such as clustering coefficient, shortest paths, network heterogeneity, the centralization, average number of neighbors and characteristic path length, showed a changed in the values of HIV-1 macrophage infected gene network, compared with that of normal macrophage (figure 6). The non-altered network was more condensed, had more number of interactions, was wide open rich in shortest paths and also was composed by one major component and two minor clusters being more heterogeneous and multi-functional table 4. Statistical differences between the topology states of two networks were registered for topological coefficients, closeness centrality and neighborhood connectivity distribuion (Kolmogorov-Smirnov test p<0.05), but not in average clustering coefficient distribution. These results indicate that normal network was significantly more central and densely connected in comparison with that of HIV-1 macrophage infected network.
Using random network plugin by Cytoscape, it was found that the clustering coefficient of the non-infected network and simulated infected network in comparison with those generated at random showed not statistical differences (Kluskal Wallis test, p=0.317). The data confirmed that the topology of both reported networks have a strong support that the simulation of our gene network is valid.
The gene ontology (GO) enrichment analysis showed that normal network was composed by 423 significant functional categories of a total of 1190. These individual significant categories could be further classified into two major groups; cell function regulation and signaling of biological process. In contrast HIV-1 infected macrophage gene network was enriched with 10 significant functional categories of a total of 40. The significantly over represented categories indicated that this emergent new gene network was composed by genes involved in metabolic process and DNA repair process.
Discussion
Data obtained from more than two thousands human genome fragments flanking lentivirus integration sites, showed a preferential distribution in medium and high multifractality chromosomes as a characteristic of the process. It could be interpreted in terms of chromatin instability as a general effect of lentivirus infection. The preferential lentiviral cDNA integration process to human chromosomes with the high and medium multifractality, significantly correlate with high density gene and Alu densities per chromosome. Therefore, characterization of the chromatin changes that occur by cDNA integration represents an important issue to understanding the complexity of crosstalk between retrovirus genome and host genome (Colin, et al, 2014).
The results also remarks the importance of the dynamic of a portion of the interphase chromatin which is far from of equilibrium or in open-decondensed chromatin regions. In this sense several variables including the nucleosomes occupancy, methylation of CpG Islands, DNase hypersen-sitive regions and transcriptionally active genes, play an important role to provide an environment for DNA regulatory processes associated with the cDNA integration such as DNA replication, repairs and transcription (Albanese, et al, 2008). We concluded that the structural characteristics and the epigenetic modifications observed in those regions with high frequency of cDNA viral integrations would synergistically configure a local complex âgenomic environ-mentâ that facilitates the target site selection during the retroviral integration in host.
As the cDNA integration do not follow a random model, some characteristics of the chromatin associated with regions of high level of lentiviral cDNA integration, support the hypothesis that a preferential integration is conditioned by structural and functional states of local chromatin. These states are defined by several genomic variables which were studied in this work, and together with others, would define local genomic environments (Alzate, et al, 2015; Craigie y Bushman, 2014; Ciuffi, et al, 2005; Ciuffi, 2008; Bushman, et al, 2005). However is important to state that beyond of the intervention of some repetitive elements such as Alu sequences, and CpG islands in a conformation of local chromatin remodeling, the multifractality of chromosomes is also a crucial variable that influence the dynamic of remodeling the local genomic environment complexity; it is a new approach that extent the analysis of complex retroviral integration process (Losa, 2009).
Previous analysis using the two-dimensional density correlation matrix, showed that all human chromosomes have common characteristics in their multifractal spectrum and deviate substantially from random and uncorrelated sequences of the same size. Small deviations are observed between the longer and the shorter chromosomes, especially for the higher (in absolute values) statistical moments (Provata and Katsaloulis, 2010). Moreover the multifractal analysis of human genome suggest that the chromosome molecular structure might be organized as a system operating far from equilibrium. In this sense, the analysis performed in the present study validated the strong relationship of lenti-virus integration process and chromosomal multifractality found especially for chromosomes 17, 19, 22, and 16.
The high multifractal chromosomes have the highest densities of Alu elements and genes per Mb (Moreno, et al, 2011). Particularly chromosomes 17 and 19 are by far, the most multifractal chromosomes have the highest gene densities of the whole genome and a high and medium frequencies of lentiviral integration. The results of the PCA support the close association between genes density/ chromosome, AvDDq and MRTR and highest multifractality values chromosomes. These associations are important to analyze in terms of chromatin stability through the cell cycle process.
Mammalian interphase nucleus is a highly organized and compartmentalized into three-dimensional discrete chromosome territories or domains (CTs) (Folle, 2008; Cremer, et al, 2008), surrounded by a network of interchromatin compartment (IC) which harbors factors involved in DNA replication or repair as well as RNA transcription and processing (Chakraborty, et al, 2015; Meaburn y Misteli, 2007; Lanctôt, et al, 2007), such organization is the scenario that required to the process of retroviral integration occur. Several studies comparing transcriptionally inactive and active chromatin found a more interior nuclear position for active chromatin mainly for chromosomes 17 and 19 (Geyer, et al, 2011; Folle, 2008; Lanctôt, et al, 2007).
Although the current knowledge shows that HIV-1 favors integration in transcribed chromosomal regions, thus improving chances for efficient expression of the viral genes for HIV-1 and HIV-2 and HIV vectors, actually it is not fully understood how the genomic site for integration is determined, (Cattoglio, et al, 2010; Felice, et al, 2009). In this sense, our data present strong evidences that one of the crucial factors to determine the integration site, among others, is the topological conformation of chromatin associated to chromosomes with high gene densities specially 19 and 17. The close association between high multifractality and high frequency of lentivirus integration in both chromosome would be dependent of the topological configuration of interphase chromatin that is the scenario for the process of lentivirus integration (García-Vallejo, et al, 2015).
Experimental results taken from human fibroblast nuclei in G0 phase showed that regarding 16, 17, 19, and 22, were located in the most internal perimeter (Bolzer, et al, 2005). In this sense, the results of the current work, could be interpreted as such 3D localization would be a result of different waves of fractal sates produced by the machinery of the preintegration complex on the structure of local associated chromatin. Taking together all sources of evidence, is possible to correlate the multifractality with the high instability of chromatin associated mainly involved in events of high rates of gene transcription as testing by the differential localization of promoters inside the nucleus.
In this study we simulated at a systemic level, the alterations of cellular pathways when HIV-1 provirus integrates into genes by turning them off and produce dysregulation of several local signaling pathways among others in MAPK and Wnt signaling process. One of the target genes associated with HIV-1 integration was AKT3, also called PKB, which is a serine/threonine protein kinase family member. It is involved in a wide range of biological processes including cell proliferation, cell differentiation, apoptosis, stimulating cell growth, and regulating other biological responses (Coffer, et al, 1998; Song, et al, 2005). Also, it has been identified playing important roles of regulation in the G2/M transition of the cell cycle (Lee, et al, 2005). According with these previous data, is possible to propose that, when AKT3 is turned off by HIV-1 integration, the cross talk with JNK, NFTA and others is disrupted leading to a signaling dysfunction of metabolic associated pathways. When AKT3 was inactive, the direct interaction with MKK7 produce a disruption of JNK and after with JUN that would result in a non-activation by phosphorilation of apoptotic and cell cycle process (Oh, et al, 2005; Cui, et al, 2008; Kowalczyk y Zablocka, 2008).
I do get strong evidence to propose that HIV-1 inte-gration in host genome disrupt several signaling pathways that control the normal cell homeostasis changing towards an anti-apoptosis gene signature associated with many hub nodes with a high degree of interactions. As HIV-1 infected macrophage is an abnormal reservoir in which the metabolic cascades are altered, it is feasible to propose that the metabolism of macrophage adapt to perform survival functions where the apoptotic process is interrupted and a SOS metabolism make that the macrophage changes its life style. Deciphering the signaling pathways involved in HIV-1 integration process in macrophage as viral reservoirs, shall be critical to a better understand HIV-1 infection for early stage viral transmission and dissemination within the host.
Studies on host genomics have revealed the existence of identifiable HIV-1 specific protective factors among infected individuals who remain naturally resistant viraemia controllers with little or no evidence of virus replication (Kaur, et al, 2013), currently, a variety of strategies are being tested in order to breakthrough this highly challenging treatment barrier. Most of them consider the genome as a target to perform either chemical modification or introduction of stable molecular devices to alter the programing of gene expression in infected host genome (Méndez, et al, 2015).
The current study provided new insights on the influence of stable retroviral integration on nuclear chromatin orga-nization and support evidence of recent studies indicating that a fractal model of chromatin architecture is consistent with structural data on chromatin interminglement, and with the diffusion and binding properties of chromatin interacting proteins (Bancaud, et al, 2012; Bancaud, et al, 2009).
It is a new approach that would contribute to open new points of view to extent the knowledge about the complex mechanisms operating during the integration of lentiviral cDNA into the genome of host cell with the aim, in a future, develop novels and powerful therapeutic strategies to control the lentivirus infection (Manjunath, et al, 2013; Bauman, et al, 2012)
Acknowledgments
I especially thanks to my associates, Doctors Martha C. Domínguez, Adalberto Sánchez, Mercedes Salcedo, Julio Cesar Montoya, José MaríaSatizábal, Pedro Moreno, Patricia Velez and Milton Quintana for supporting different activities of the research group LABIOMOL group A Colciencias 2015. To my friends and colleagues Max Essex, School of Public Health of Harvard University; King Jordan and Fred Vannenberg, Georgia Technology Institute, and Leonardo Mariño of NCBI/NIH. To my graduated students and members of LABIOMOL: Juliana Soto, Ángela Peña, Dianora Fajardo, Alejandra Rodríguez, Lina Alzate, Meliza Santiago, Paola Andrea Lucumí, and Maria Alejandra Abonia. Especially thanks to those financing agencies that support the studies on retrovirus integration: Fogarty/ NIH, Fullbright, Georgia Technology Institute, Ministry of Culture and Education of Japan, Colciencias, FundaciónBanco de la República, Colombia. To the Universidad del Valle, my scientific home, for financing and supporting several scientific projects of my group.
Conflict of interest
Author declare do not have any conflict of interest about the content of the article
References
Albanese A, Arosio D, Terreni M, Cereseto A. 2008. HIV-1 Pre- Integration Complexes Selectively Target Decondensed Chromatin in the Nuclear Periphery. PLoS one. 3: e2413. doi:10.1371/journal.pone.0002413. [ Links ]
Alzate LA, Domínguez MC, Sánchez A, Vélez P, Moreno PA, García-Vallejo F. 2015. Eventos fractales relacionados con fenómenos epigenéticos del genoma humano en la integración de los lentivirus humanos. Biomédica. 35: 53. [ Links ]
Balakrishnan S. 2009. Alternative paths in HIV-1 targeted human signal transduction pathways. BMC Genomics. 10 (Suppl 3):S30. [ Links ]
Bancaud A, Huet S, Daigle N, Mozziconacci J, Beaudouin J, Ellenberg J. 2009. Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organization of chromatin. EMBO J. 28 (24): 3785-98. [ Links ]
Bancaud A, Lavelle C, Huet S, Ellenberg J. 2012. A fractal model for nuclear organization: current evidence and biological implications. Nucleic Acids Res. 40 (18): 8783-92. [ Links ]
Barr S, CiuffiA, Leipzig J, Shinn P, Ecker J, Bushman F. 2006. HIV Integration site selection: targeting in macrophages and the effects of different routes of viral entry. Mol. Ther. 14: 218-25. [ Links ]
Benleulmi MS, Matysiak J, Henriquez DR, Vaillant C, Lesbats P, Calmels C, Naughtin M, et al. 2015. Intasome architecture and chromatin density modulate retroviral integration into nucleosome. Retrovirology. 12: 13. [ Links ]
Bolzer A, Kreth G, Solovei I, Koehler D, Saracoglu K, Fauth C, Müller S. 2005. Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 3 (5): e157. [ Links ]
Bushman F, Lewinski M, Ciuffi A, Barr S, Leipzig J, Hannenhalli S, Hoffmann C. 2005. Genome-wide analysis of retroviral DNA integration. Nature Rev. Microbiol. 3: 848-58. [ Links ]
Carteau S, Hoffmann C, Bushman F. 1998. Chromosome struc-ture and human immunodeficiency virus type 1 cDNA integration: centromeric alphoid repeats are a disfavored target. J. Virol. 72: 4005-4014. [ Links ]
Cattoglio C, Pellin D, Rizzi E, Maruggi G, Corti G, Miselli F, Sartori D, et al. 2010. High-definition mapping of retroviral integration sites identifies active regulatory elements in human multipotent hematopoietic progenitors. Blood. 116 (25): 5507-17. [ Links ]
Cereseto A, and Giacca M. 2004. Integration site selection by retroviruses. AIDS Rev. 6: 13-21. [ Links ]
Chakraborty S, Mehta I, Kulashreshtha M, Rao BJ. 2015. Quantitative analysis of chromosome localization in the nucleus. Methods Mol Biol. 1228: 223-33. [ Links ]
Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, et al. 2005. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 11: 1287-89. [ Links ]
Ciuffi A. 2008. Mechanisms governing lentivirus integration site selection. Curr GeneTher. 8: 419-29. [ Links ]
Coffer PJ, Jin J, Woodgett JR, 1998. Protein kinase B (c-Akt): a multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem J. 335 (1): 1-13. [ Links ]
Coffin JM. Retroviridae and their replication In Virology, 1996. ed. BN Fields et al., Raven Press, New York. pp. 1767-1848. [ Links ]
Colin L, Verdin E, Van Lint C. 2014. HIV-1 Chromatin, Transcription, and the Regulatory Protein Tat. Methods. Mol Biol. 1087: 85-101. [ Links ]
Craigie R, Bushman FD. 2014. Host Factors in Retroviral Integration and the Selection of Integration Target Sites. Microbiol Spectr. 2 (6). doi: 10.1128. [ Links ]
Cremer M, Grasser F, Lanctot C, Muller S, Neusser M, Zinner R, Solovei I, et al. 2008. Multicolor 3D fluorescence in situ hybridization for imaging interphase chromosomes. Methods Mol Biol. 463: 205-3. [ Links ]
Crise B, Li Y, Yuan C, Morcock DR, Whitby D, Munroe DJ, Arthur LO, et al. 2005. Simian immunodeficiency virus integration preference is similar to that of human immuno-deficiency virus type 1. J Virol. 79: 12199-204. [ Links ]
Cui M, Huang Y, Zhao Y, Zheng J. 2008. Transcription factor FOXO3a mediates apoptosis in HIV-1-infected macro-phages. J. Immunol. 15 (2): 898-906. [ Links ]
DeCerbo J, Carmichael GG. 2005. SINEs point to abundant editing in the human genome. Genome Biology. 216: 1-4. [ Links ]
Demeulemeester J, De Rijck J, Gijsbers R, Debyser Z. 2015 Retroviral integration: Site matters: Mechanisms and conse-quences of retroviral integration site selection. Bioessays. 37 (11): 1202-14. [ Links ]
Derse D, Crise B, Li Y, Princler G, Stewart C, Connor F, Hughes H, et al. 2007. HTLV-1 integration target sites in the human genome: comparison with other retroviruses. J. virol. 81: 6731-6741. [ Links ]
Diehl AG, Boyle AP. 2016. Deciphering ENCODE. Trends Genet. 32 (4): 238-49. [ Links ]
The ENCODE Project Consortium. 2012. Nature. 489: 57-74. [ Links ]
ENCODE Project Consortium. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 447 (7146): 799-816. [ Links ]
Felice B, Cattoglio C, Cittaro D, Testa A, Miccio A, Ferrari G, Luzi L, et al. 2009. Transcription factor binding sites are genetic determinants of retroviral integration in the human genome. PLoS One. 4 (2): e4571. [ Links ]
Ferris AL, Wu X, Hughes CM, Stewart C, Smith SJ, Milne TA, Wang GG, et al. 2010. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc. natl. acad. sci. USA. 107: 3135-40. [ Links ]
Folle GA. 2008. Nuclear architecture, chromosome domains and genetic damage. Mutat Res. 658 (3): 172-83. [ Links ]
García-Vallejo F, Domínguez MC, Sánchez A, Vélez P, Moreno P. 2015. Integración preferencial del ADN complementario de los lentivirus humanos en cromosomas con alto nivel de multifractalidad. Biomédica. 35: 54. [ Links ]
Geyer PK, Vitalini MW, Wallrath LL. 2011 Nuclear organization: taking a position on gene expression. Curr Opin Cell Biol. 23 (3): 354-9. [ Links ]
Folle GA. 2008. Nuclear architecture, chromosome domains and genetic damage. Mutat Res. 658 (3): 172-83. [ Links ]
Hematti P, Hong BK, Ferguson C, Adler R, Hanawa H, Sellers S, Ingeborg E. 2004. Distinct Genomic Integration of MLV and SIV Vectors in Primate Hematopoietic Stem and Progenitor Cells. PLoS Biol. 2: E423. [ Links ]
Hindmarsh P, Leis J. 1999. Retroviral DNA integration. Microbiol mol. biol. rev. 63: 836-84. [ Links ]
Ideker T, Ozier O, Schwikowski B, Siegel AF. 2002. Discovering regulatory and signaling circuits in molecular interaction networks. Bioinformatics. 18: S233-S240. [ Links ]
Ikeda T, Shibata J, Yoshimura K, Koito A, Matsushita S. 2007. Recurrent HIV-1 integration at the BACH2 locus in resting CD4+ T cell populations during effective highly active antiretroviral therapy. J Infect Dis. 195 (5): 716-25. [ Links ]
International human genome sequencing consortium. 2004. Finishing the euchromatic sequence of the human genome. Nature. 431: 931-45. [ Links ]
Jordan A, Defechereux P, Verdin E. 2001.The site of HIV- 1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. The EMBO J. 20: 1726-38. [ Links ]
Karimi-Busheri F, Rasouli-Nia A. 2015. Integration, Networking, and Global Biobanking in the Age of New Biology. Adv Exp Med Biol. 864: 1-9. [ Links ]
Kaur G, Sharma G, Kumar N, Kaul MH, Bansal RA, Vajpayee M, Wig N. et al. 2013. Genomic architecture of HIV-1 infection: current status & challenges. Indian J Med Res. 138 (5): 663-81. [ Links ]
Klug M, Heinz S, Gebhard C, Schwarzfischer L, Krause SW, Andreesen R, Rehli M. 2010. Active DNA demethylation in human postmitoticcells correlates with activating histone modifications, but not transcription levels. Genome Biol. 11 (16): R63. [ Links ]
Kowalczyk JE, Zablocka B. 2008. Protein kinases in mitochondria. Postepy Biochem. 54: 209-16. [ Links ]
Lanctôt C, Cheutin T, Cremer M, Cavalli G, Cremer T. 2007. Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat Rev Genet. 8 (2): 104-15. [ Links ]
Le Sage V, Mouland AJ, Valiente-Echeverría F. 2014. Roles of HIV-1 capsid in viral replication and immune evasion. Virus Res. 193: 116-29. [ Links ]
Lee SR, Park JH, Park EK, Chung CH, Kang SS, Bang OS. 2005. Akt-induced promotion of cell-cycle progression at G2/M phase involves upregulation of NF-Y binding activity in PC12 cells. J. Cell. Physiol. 205 (2): 270-77. [ Links ]
Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, et al. 2007. The diploid genome sequence of an individual human. PLoS Biol. 5: e254. [ Links ]
Lewinski M, Yamashita M, Emerman M, Ciuffi A, Marshall H, Crawford G. 2006. Retroviral DNA Integration: Viral and Cellular Determinants of Target-Site Selection. PLoS pathog. 2: 0611-0622. [ Links ]
Losa GA. 2009.The fractal geometry of life. Riv Biol. 102 (1): 29-59. [ Links ]
MacNeil A, Sankale JL, Meloni S, Sarr A, Mboup S, Kanki P. 2006. Genomic Sites of Human Immunodeficiency Virus Type 2 (HIV-2) Integration: Similarities to HIV-1 In Vitro and Possible Differences In Vivo. J. virol. 80: 7316-21. [ Links ]
Maere S, Heymans K, Kuiper M. 2005. BiNGO: a cytoscape plug-in to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21 (16): 3448-49. [ Links ]
Manjunath N, Yi G, Dang Y, Shankar P. 2013. Newer gene editing technologies toward HIV gene therapy. Viruses. 5 (11): 2748-66. [ Links ]
Bauman JD, Patel D, Arnold E. 2012. Fragment screening and HIV therapeutics. Top Curr Chem. 317: 181-200. [ Links ]
Margolis DA, Boffito M. 2015. Long-acting antiviral agents for HIV treatment. Curr Opin HIV AIDS. 10 (4): 246-52. [ Links ]
Maxfield L, Fraize C, Coffin JM. 2005. Relationship between retroviral DNA-integration site selection and host cell transcription. Proc natl. acad. Sci USA. 102: 1436-44. [ Links ]
Mcsweeney. Google Summer of Code 2008. http://sites.google.com/site/randomnetworkplugin/. [ Links ]
Meaburn KJ, Misteli T. Cell biology: chromosome territories. Nature. 2007; 445 (7126): 379-81. [ Links ]
Méndez C, Ahlenstiel CL, Kelleher AD. 2015. Post-transcriptional gene silencing, transcriptional gene silencing and human immunodeficiency virus. World J Virol. 4 (3): 219-44. [ Links ]
Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, Ecker JR, et al. 2004. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2 (8): E234. [ Links ]
Moore J.P, Stevenson M. 2000. New Targets for Inhibitors of HIV-1 Replication. Nature Rev. 1: 40-9. [ Links ]
Moraes F, Góes A. 2016. A decade of human genome project conclusion: Scientific diffusion about our genome knowl-edge. Biochem Mol Biol Educ. Mar 7. doi: 10.1002/ bmb.20952. [ Links ]
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, et al. 2001. Initial sequencing and analysis of the human genome. Nature. 409: 860-921. [ Links ]
Moreno PA, Vélez PE, Martínez E, Garreta LE, Díaz N, Amador S, Tischer I, et al. 2011. The human genome: a multifractal analysis. BMC Genomics. 12: 506. doi: 10. 1186/1471-2164-12-506. [ Links ]
Oh SW, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis R, Tissenbaum HA, Piot P, UNAIDSâLancet Commission. 2015. Defeating AIDS advancing global health. Lancet. 386: 171-218. [ Links ]
Polychronopoulos D, Athanasopoulou L, Almirantis Y. 2016 Fractality and entropic scaling in the chromosomal distribution of conserved noncoding elements in the human genome. Gene. Feb 17. pii: S0378-1119. [ Links ]
Provata A, Katsaloulis P. 2010. Hierarchical multifractal represen-tation of symbolic sequences and application to human chromosomes. Phys Rev E Stat Nonlin Soft Matter Phys. 81: 26-102. [ Links ]
Rick SM, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, Ecker JR, et al. 2004. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2: 1127-37. [ Links ]
Saayman S, Ali SA, Morris KV, Weinberg MS. 2015. The therapeutic application of CRISPR/Cas9 technologies for HIV. Expert Opin Biol Ther. 15 (6): 819-3. [ Links ]
Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov W, Howe V, Li J, et al. 2006. TM4 microarray software suite. Methods Enzymol. 411: 134-193. [ Links ]
Schröder A, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. 2002. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell. 110: 521-29. [ Links ]
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N. 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13 (11), 2498-2504. [ Links ]
Sierra S, Kupfer B, Kaiser R. 2005. Basics of the virology of HIV-1 and its replication. J. Clin. Virol. 34: 233-44. [ Links ]
Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, et al. 2006. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nature Gen. 38: 1348-54. [ Links ]
Song G, Ouyang G, Bao S. 2005. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 9 (1): 59-71. [ Links ]
Soto J, Peña A, Salcedo M, Domínguez MC, Sánchez A, García-Vallejo F. 2010. Caracterización Genómica de la Integración In vitro del VIH-1 en células mononucleares de sangre periférica, macrófagos, y células T de Jurkat. Infectio. 14: 20-30. [ Links ]
Soto J, Peña A, García-Vallejo F. 2011. A genomic and bioinformatics analysis of the integration of HIV in peripheral blood mononuclear cells. AIDS Res Hum Retroviruses. 27: 547-55. [ Links ]
Soto-Girón MJ, García-Vallejo F. 2012. Changes in the topology of gene expression networks by human immunodeficiency virus type 1 (HIV-1) integration in macrophages. Virus Res. 163 (1): 91-7. [ Links ]
UNAIDS JUNPoHA Global Report: UNAIDS report on the global AIDS epidemic. 2013 [ Links ]
Van Maele B, Busschots K, Vandekerckhove L, Christ F, Debyser Z. 2006. Cellular co-factors of VIH-1 integration. Trends Biochem. Sci. 31: 98-105. [ Links ]
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, et al. 2001. The sequence of the human genome. Science. 291: 1304-51. [ Links ]
Versteeg R, van Schaik BDC, van Batenburg MF, Roos M, Monajemi R, Caron H, Bussemaker HJ, et al. 2003. The human transcriptome map reveals extremes in gene density, intron length, GC content, and repeat pattern for domains of highly and weakly expressed genes. Genome Research. 13: 1998-2004. [ Links ]
Wang YJ, McKenna PM, Hrin R, Felock P, Lu M, Jones KG, Coburn CA, Grobler JA. 2010. Assessment of the susceptibility of mutant HIV-1 to antiviral agents. J. virol methods. 165: 230-7. [ Links ]
Wong RW, Mamede JI, Hope TJ. 2015. Impact of Nucleoporin- Mediated Chromatin Localization and Nuclear Architecture on HIV Integration Site Selection. J Virol. 89 (19): 9702-5. [ Links ]
Wu X, Li Y, Crise B, Burgess SM. 2003. Transcription start regions in the human genome are favored targets for MLV integration. Science. 300: 1749-51. [ Links ]
Yi J, Stypula-Cyrus Y, Blaha CS, Roy HK, Backman V. 2015. Fractal Characterization of Chromatin Decompaction in Live Cells. Biophys J. 109 (11): 2218-26. [ Links ]
Zaccarelli M, Tozzi V, Lorenzini P, Trotta MP, Forbici F, Visco-Comandini U, Gori C, et al. 2005. Multiple drug class-wide resistance associated with poorer survival after treatment failure in a cohort of HIV-infected patients. AIDS. 19: 1081-89. [ Links ]
