










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista de la Academia Colombiana de Ciencias Exactas, Físicas y Naturales
Print version ISSN 0370-3908
Rev. acad. colomb. cienc. exact. fis. nat. vol.40 no.157 Bogotá Oct./Dec. 2016
https://doi.org/10.18257/raccefyn.381
doi: http://dx.doi.org/10.18257/raccefyn.381
1 Grupo de Microbiología Clínica y Ambiental, Programa de Biología, Facultad de Ciencias Exactas y Naturales, Universidad de Cartagena, Campus San Pablo, Cartagena, Colombia. *Correspondencia: Rosa Baldiris-Avila, rbaldirisa@unicartagena.edu.co
2 Grupo de investigación CIPTEC, Programa de ingeniería de procesos, Fundación Universitaria Tecnológico Comfenalco, Cartagena, Colombia
3 Grupo de Química Cuántica y Teórica, Facultad de Ciencias Exactas y Naturales, Universidad de Cartagena, Campus San Pablo, Cartagena, Colombia
Recibido: 25 de mayo de 2016. Aceptado: 23 de noviembre de 2016
Resumen
El síndrome de Lowe (SL) es una enfermedad transmitida por herencia ligada al sexo, que afecta mayoritariamente a los hombres y muy ocasionalmente a las mujeres. El SL consiste en mutaciones en el gen OCRL1 que afectan el metabolismo del inositol fosfato, produciendo afecciones significativas de los ojos, el sistema nervioso y los riñones. En este estudio se construyó una red de interacción proteína-proteína para la enzima fosfatidilinositol 4,5-bifosfato 5-fosfatasa, en el software Cytoscape versión 3.3.0, a partir de información suministrada por las bases de datos KEGG, PubMed, BioGRID, STRING y Uniprot, donde se extrajeron 26 proteínas relacionadas con OCRL. Con base en un análisis funcional y topológico, se obtuvo que las categorías ontológicas (GO) de mayor significancia están relacionadas con el transporte mediado por vesículas y la transducción de señales medidas por pequeñas GTPasas. Por otro lado, los parámetros topológicos de la red construida fueron, coeficiente de transitividad: 0,033, diámetro: 5,0 centralización: 0,55.
Palabras Clave: Síndrome de Lowe; Cytoscape 3.3.0; OCRL; Bioinformática; PPIs.
Abstract
Lowe syndrome is a disease transmitted by sex-linked inheritance. That mainly affects males, and very occasionally women. It consists of a mutation in the gene OCRL1 that affects inositol phosphate metabolism, producing significant problems on the eye, nervous system and kidneys. A network of protein-protein interaction for the enzyme phosphatidylinositol 4,5-bisphosphate 5-phosphatase in the Cytoscape software version 3.3.0 was build, from the information provided by the KEGG database, PubMed, BioGrid, STRING, Uniprot data, 26 proteins were extracted related with OCRL. Based on a functional and topological analysis, was obtained ontological categories (GO) of greater significance are related mediated transport vesicles and signal transduction steps by small GTPases. On the other hand, topological parameters of the network were built, transitivity coefficient: 0.033, diameter: 5,0 centralization: 0.55.
Key words: Lowe syndrome; Cytoscape 3.3.0; OCRL; Bioinformatics; PPIs.
Introducción
El síndrome de Lowe o distrofia óculo-cerebro-renal es una enfermedad hereditaria transmitida por herencia recesiva ligada al cromosoma X, por lo que afecta en la mayoría de los casos a los hombres; las anomalías genéticas relacionadas con este síndrome se producen por deleciones, translocaciones y mutaciones stop, con una fracción más pequeña que ocurre como splicing y mutaciones sin sentido (Pirruccello & De Camilli, 2012). Ocasionalmente se han reportado algunos casos en que este síndrome se expresa en las mujeres, que por lo general, se explica por hipótesis de Lyon o por heterogeneidad genética. Sin embargo, estudios recientes han demostrado que la causa más probable de la aparición en mujeres es una translocación del segmento del cromosoma X que contiene el gen, a un autosoma con inactivación preferencial de la X íntegra (Avendaño, et al., 2009).
Se considera una enfermedad rara, debido a que se estima que su incidencia en la población es de 1 por cada 500.000 recién nacidos (Kornfeld, Synder, MacGee, Russell, Appenzeller, 1975). El reporte de caso más reciente para Latinoamérica fue publicado por Casilla, et al., (2016) en un varón de 55 años de edad, con fuertes afecciones oculares y renales; a quien se le realizó luego de su diagnóstico un tratamiento médico y quirúrgico. Sin embargo, debido a las consecuencias letales de la enfermedad, el pronóstico fue reservado. En Colombia hasta la fecha, segundo trimestre de este año, de acuerdo a la información suministrada por la Federación Colombiana de Enfermedades Raras (FECOER, http://www.fecoer.org/), no hay casos de pacientes con síndrome de Lowe reportados.
El síndrome óculo-cerebro-renal, consiste en un trastorno del metabolismo del inositol fosfato, que desempeña funciones reguladoras importantes en la fisiología celular, provocando alteraciones significativas en los ojos (cataratas y glaucomas congénitos), el sistema nervioso (arreflexia, retardo mental, hipotonía neonatal) y disfunción renal túbulo proximal (síndrome de Fanconi) que se caracteriza por el deterioro de la reabsorción de las proteínas de bajo peso molecular, iones y metabolitos (McCrea, et al., 2006), en algunos casos se puede presentar comportamiento agresivo y dimorfismos faciales (Avery, et al., 2001; Nussbaum & Suchy, 2001; Charnas, et al., 1991). Aproximadamente alrededor de la mitad de los pacientes presentan convulsiones (Fejerman & Fernández, 2007).
Este síndrome se produce por una mutación en el gen OCRL1, el cual ha sido localizado en la región Xq25-q26; el producto proteico de OCRL1 es la enzima fosfatidilinositol 4,5-bifosfato 5-fosfatasa, que posee 901 aminoácidos y un peso de 105 kDa.
La enzima fosfatidilinositol 4,5-bifosfato 5-fosfatasa reside en una serie de estructuras vesiculares en todo el sistema endosomal y el complejo de Golgi. También está presente en la membrana plasmática en ondulaciones de la membrana y en depresiones revestidas de clatrina; esta amplia distribución esta mediada por diversas interacciones: une clatrinas, adaptadores de clatrina, varias GTPasas, y la proteína endocítica APPL1.
En su estructura OCRL1 posee cuatro dominios: PH (dominio con homología a Pleckstrina), RhoGAP (Proteina activadora Rho GTPasa), 5-fosfatasa y ASH (ASPM-SPD2- Hidina) (Madhivanan, Ramadesikan & Aguilar, 2015), dos cajas de clatrina (Pirruccello & De Camilli, 2012), que están implicadas en la polimerización de actina, el transporte de proteínas y la señalización celular (Krauβ & Haucke, 2007). Se han relacionado dos fenotipos distintos a nivel celular para este síndrome implicados en la remodelación de la membrana y los cilios primarios, los cuales se hayan vinculados a defectos en el dominio RhoGAP (Conduit, Dyson, Mitchell, 2012).
Debido a que la enfermedad esta categorizada como rara y que la expectativa de vida promedio de los pacientes es bastante baja, alrededor de 30 a 40 años en general, es importante el desarrollo de estudios encaminados hacia la creación de tratamientos que permitan mejorar tanto las condiciones de salud como aumentar la funcionalidad de los pacientes. En la actualidad, se ha propuesto la cirugía de cataratas, el control de glaucoma, la terapia física y de lenguaje, el uso de medicamentos para la conducta, la corrección de la acidosis tubular y alteraciones óseas mediante la utilización de bicarbonato, fosfato y potasio, como posibles tratamientos para esta enfermedad (Casilla, Villar, Lizárraga, 2016).
El propósito de este trabajo fue construir y analizar una red interacciones proteína-proteína de la fosfatidilinositol 4,5-bifosfato 5-fosfatasa, relacionada con las rutas metabólicas afectadas en el síndrome de Lowe.
Materiales y métodos
La metodología en este estudio comprendió dos fases. En la fase inicial o búsqueda de información, se obtuvieron los síndromes relacionados con la enfermedad de Lowe en las bases de datos: PubMed (http://www.ncbi.nlm.nih.gov/pubmed/, DynaMED (http://www.dynamed.com/home/), Science direct (http://www.sciencedirect.com/), las rutas metabólicas fueron consultadas en la base de datos KEGG (http://www.genome.jp/kegg/) (Kanehisa, 2012), y las proteínas que interactúan con OCRL en Uniprot (http://www.uniprot.org/), BioGRID (http://thebiogrid.org) (Chatr-Aryamontri, et al., 2014) y STRING (http://string-db.org) (Szklarczyk, et al., 2015). Tomando como dato de entrada OCRL y el organismo Homo sapiens (Human). Seguidamente se compararon las interacciones encontradas en las distintas bases de datos, para la selección de los nodos de la red.
En la segunda fase, con las 27 proteínas extraídas (NTRK1, ELAVL1, RAB5A, GTSE1, OCRL, NXF1, GRB2, CLTC, RAB8A, RAB1A, RAB6A, SSX2IP, XPO1, FAM109A, FAM109B, SPINT2, GOLGA5, CEP128, SCLT1, CEP170, HERC2, SCN2B, ENPP6, VSIG4, BTN2A1, CACNG4, RAB14), se construyó una red de interacción en el Software Cytoscape versión 3.3.0 (Shannon, et al., 2003). Para ello, se descargaron las interacciones de OCRL registradas en BioGRID, así como las interacciones de sus nodos adyacentes. Cabe resaltar que en el archivo descargado, no se presentan interacciones entre las proteínas OCRL y APPL1. Sin embargo, la literatura citada (Pirruccello & De Camilli, 2012) describe la interacción entre estas moléculas. sPor ello, este nodo y sus interacciones adyacentes se tuvieron en cuenta para la construcción de la red.
Se ingresaron los archivos extraídos de la base de datos BioGRID a Cytoscape; con la función Merge de NetworkAnalyzer se construyó la red usando la opción unión, seleccionando todas las redes ingresadas individualmente. Posteriormente, se realizó un análisis funcional estableciendo las categorías ontológicas (en adelante, GO) de mayor significancia, aplicando un test hipergeométrico (p<0,005), ajustado con corrección Bonferroni Family-Wise. Para los archivos ontológicos: funciones moleculares, procesos biológicos y componentes celulares; en el plug-in GOlorize. Con base en lo cual se definieron aspectos estéticos de la red, como lo son el diseño, estilo, colores de los nodos y tamaño de los mismos.
Por otra parte, se utilizó el complemento Network Analyzer para determinar los parámetros tipológicos de la red, seleccionando todos los nodos y la opción Analysis Network. El parámetro Degree o número de enlaces fue escogido como atributo de la red. Con la opción Generar estilo de estadísticas, se seleccionó la escala cromática de verde a rojo, siendo estos los menores y mayores valores de Degree respectivamente.
Resultados y discusión
En este estudio se aplicaron herramientas bioinformáticas que permitieron condensar una serie de hallazgos importantes relacionados con la enfermedad de Lowe. Sin embargo, la fisiopatología del síndrome permanece en gran parte inexplicada, a pesar del conocimiento de la disfunción enzimática existente y de que el gen OCRL1 se expresa en numerosos tejidos (Avendaño, et al., 2009).
OCRL y el Sindrome de Lowe. La enzima fosfatidilinositol 4,5-bifosfato 5-fosfatasa posee dos dominios (PH y Rho GAP), dos regiones (ASH, 5-fosfatasa) y dos motivos de clatrina. El dominio PH se conoce por su capacidad para unirse a fosfoinosítidos con alta afinidad y especificidad, aunque actualmente se ha demostrado que menos del 10% de los dominios PH poseen esta propiedad. Los casos en los que los dominios PH se unen con alta afinidad y específicamente a fosfoinosítidos están restringidos únicamente a aquellos fosfoinosítidos que tienen un par de fosfatos adyacentes en su inositol en la posición inicial (Headgroup) (Figura 4). El 90% de los dominios PH que no poseen la capacidad de unirse fuertemente y específicamente a fosfoinosítidos, han sido poco estudiados (Lemmon, 2007).
El dominio Rho es una subfamilia que comprende a Rho A, B, C, D y E. Todas las proteínas G tienen secuencias consenso de aminoácidos para interacciones especificas con GDP, GTP y para la actividad GTPasa, además de tener regiones que interactúan con efectores descendentes. Estas proteínas tienen secuencias Cis-A-AX en la región C-terminal a la que se unen grupos fernesilo, geranilogeranilo, palmítico o metilo necesarios para la unión de las pequeñas proteínas G a la membrana. Estas proteínas tienen dos formas interconvertibles: la forma unida a GTP o activa y la unida a GDP o inactiva, el paso de una forma a otra es facilitados por dos tipos de proteínas reguladoras, la GEFs y GDI. Este dominio está implicado en varios procesos celulares como organización del citoesqueleto, tráfico de membranas, crecimiento celular, transcripción de genes, entre otros (Taleisnik, 2006).
La relación entre OCRL y la formación de los cilios, sugiere una implicación directa en la patología de esta enfermedad. Estudios realizados en pacientes con este síndrome, demuestran una desmielinización que puede ser una consecuencia del papel que juega OCRL en el tráfico de megalina en los riñones, puesto que las megalinas unen los ligados de la vía de señalización de los cilios, median la endocitosis y la señalización corriente abajo, de este modo los defectos en el tráfico de megalina podrían explicar las afecciones renales del síndrome (Conduit, Dyson, Mitchell, 2012).
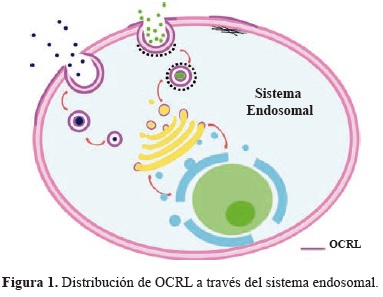
OCRL está presente en membranas de endosomas y del aparato de Golgi y se presume que su deficiencia puede afectar el tráfico de proteínas (Figura 1). La polimerización de la actina esta también regulada por OCRL, cuya integridad es necesaria tanto para la función tubular proximal como para la diferenciación del cristalino (Avendaño, et al., 2009).
Síndrome de Lowe y su relación con otras enfermedades. A pesar de que se trata de una enfermedad causada por mutaciones relacionadas a una proteína en particular, estas mutaciones se ven reflejadas en la agrupación de diferentes padecimientos en distintos tejidos. Algunos de ellos similares a los que presentan pacientes con otras enfermedades. En la Tabla 1, se relacionan las características generales de algunas enfermedades que comprometen el correcto funcionamiento del sistema excretor, específicamente afecciones renales.
En particular, la enfermedad de Dent 2, que también es ocasionada por mutaciones en el gen OCRL1. Sin embargo, esta enfermedad suele comprometer únicamente a los riñones, mientras que los pacientes con el síndrome de Lowe, pueden presentar, además de problemas en los riñones, opacidad en el cristalino, disminución en el tono muscular y depresiones severas del sistema nervioso. Estas últimas se ven reflejadas en el fenotipo característico de pacientes con síndrome óculo-cerebro-renal.
En la Tabla 2 se describen algunas de las características generales de las enfermedades que están relacionadas con el síndrome de Lowe debido a la expresión fenotípica del mismo, como lo son la deficiencia en el sentido de la vista, en función de la presencia de una nube u opacidad en el cristalino. Frente a esta dificultad se ha propuesto como tratamiento la cirugía, sin embargo, los pacientes deben continuar en un constante chequeo debido a que el éxito de la misma es bastante incierto. Por otro lado, el aumento en el tamaño de los ojos característico en pacientes con glaucoma que además está acompañado de lagrimeo excesivo; la disminución en el tono muscular propia en los primeros meses de vida y las dificultades en el sistema nervioso que se ven reflejadas en retardo mental.
Algunas mutaciones del gen OCRL1 relacionadas con el síndrome de Lowe. Hasta la fecha, han sido descritas algunas de las interacciones que presenta la enzima fosfatidilinositol 4,5-bisfosfato 5-fosfatasa. Entre ellas, su interacción con la proteína APPL1 a través del dominio RhoGAP que posteriormente es sustituida por FAM109A Y FAM109B en endosomas maduros (Mao, et al., 2009). Las mutaciones sin sentido encontradas en pacientes con Lowe, en la región COOH terminal de OCRL, no afectan la unión de esta a proteínas Rac y clatrina, pero interrumpen la interacción con APPL1 (McCrea, et al., 2008).
Las mutaciones sin sentido o terminadoras que se presentan en el dominio RhoGAP, desestabilizan la proteína afectando la formación del motivo de unión F y H de OCRL que depende crucialmente del plegamiento de la misma, lo cual no permite la unión del motivo, provocando diferentes efectos sobre las interacciones de Rho y Rab (GTPasas), sin tener efecto sobre las interacciones de clatrina. Sin embargo, en algunos pacientes con síndrome de Lowe, estas mutaciones no desestabilizan la proteína pero si afectan la unión a Rab, debido a que en este tipo de mutaciones se sustituye una base por otra, esta sustitución produce un codón de terminación que da como resultado una proteína acortada, puesto que la traducción no progresa más allá del punto donde ocurrió la mutación (Sadava, 2009) y las deleciones e inserciones identificadas en pacientes con síndrome de Lowe demuestran la importancia de las interacciones de OCRL en su función fisiológica (Pirruccello & De Camilli, 2012).
Por otra parte, en algunos estudios se han encontrado mutaciones en el dominio PH de OCRL, codificado por los exones 2-5 del gen; en este caso, se afectan los sitios de unión de clatrina. En cuanto a la región 5-fosfatasa, en el trabajo realizado por Pirruccello & De Camilli (2012) se relacionan mutaciones en pacientes con esta enfermedad, las cuales afectan directamente el plegamiento de la proteína o la unión del sustrato.
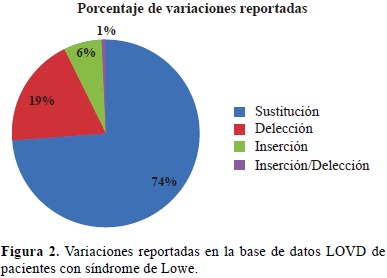
En la Figura 2 se presentan las variaciones reportadas en la base de datos Leiden Open Variation Database (LOVD, www.ncbi.nlm.nih.gov/lovd/home.php) de NCBI de pacientes con síndrome de Lowe, donde el 74% de las mismas corresponde a sustituciones de bases nitrogenadas y el 91% de ellas se han dado en la región codificante o exones del gen. Así mismo, Las variaciones por deleciones e inserciones se han encontrado solo en las regiones codificantes.
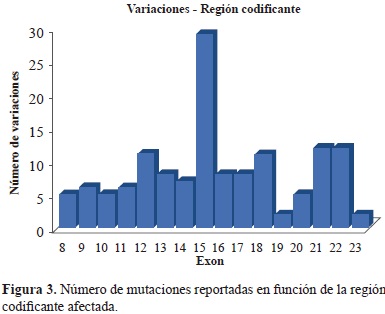
Con un total de 137 variaciones en la secuencia de este gen, el 93% de las mismas corresponden a mutaciones en regiones codificantes. En la Figura 3 se relacionan el número de variaciones reportadas por cada exón afectado del gen OCRL1, donde el exón 15 presenta la mayor frecuencia de mutación. La mutación de sentido erróneo 1499G>A (R500Q) posee la mayor frecuencia de registro para este exón, puesto que de acuerdo a la base de datos LOVD ha sido 7 veces reportada. Esta mutación consiste en un cambio en la posición 1499 de la secuencia codificante que resulta en una sustitución de arginina por glutamina. Se presume que es causada por una desaminación espontánea de una 5-metilcitosina (Lin, et al., 1998), en este caso la pérdida del grupo amino convierte a la 5-metilcitosina en una timina y luego de la replicación la hebra nueva tiene una A en lugar de una G, cambiando el codón CGA de arginina al codón CAA de glutamina, en la posición 500 de la secuencia de aminoácidos de OCRL correspondiente a la región 5-fosfatasa, que como se mencionó anteriormente puede repercutir directamente sobre el plegamiento de la proteína o dificultar la unión al sustrato, puesto que el cambio físicamente se traduce en una sustitución de un residuo grande y básico, por un residuo mediano de carácter polar. Se cree que esta mutación puede ser una de las causantes de la expresión del fenotipo de esta enfermedad (Chen, 2014).
Sin embargo, no todas las mutaciones que se presentan en OCRL1 dan paso a esta patología. La enfermedad de Dent 2, es considerada una variante leve de este síndrome, ocasionada por mutaciones en OCRL1, y claramente distinta de Dent, que es causada por mutaciones en el gen CLCN5 que codifica el canal transportador de cloruro, el cual no presenta variaciones en pacientes con afecciones renales de tipo Dent 2 (Shrimpton, et al., 2009).
Las mutaciones reportadas que causan la enfermedad de Dent 2 se encuentran principalmente en la sección 5' del gen. Están relacionadas con deleciones y mutaciones en el marco de lectura. La mutación c.833_838del (Glu278_Leu279del) provoca la eliminación del ácido glutámico y la leucina en las posiciones 278 y 279 de la proteína. Estos aminoácidos hacen parte de una región conservada en dominios de la familia endonucleasa/exonuclasa/fosfatasa en humanos. El residuo de Glu en la posición 278 es el responsable de la unión a Mg2+, un cofactor importante en la actividad catalítica de OCRL, dado que está implicado en la interacción de la enzima con su sustrato. Por otro lado, el residuo de leucina, ubicado en la posición 279, participa en las interacciones hidrofóbicas dentro de la estructura normal de OCRL. Por tanto, la presencia de esta mutación compromete la actividad enzimática y la estabilidad de la proteína (Li, et al., 2016). Lo que explicaría, la acumulación de fosfatidilinositol 4,5-bifosfato en las células y defectos en la distribución de alfa-actina, F-actina y la ciligenesis principalmente en la alteración de la formación de los cilios primarios (CP) (Madhivanan, et al., 2015); que son extensiones dinámicas de la membrana que poseen características muy específicas y su formación al igual que su mantenimiento dependen del tráfico vesicular (Madhivanan & Aguilar, 2014). Estas estructuras están presentes de manera semipermanente en muchas células diferenciadas, incluyendo las células epiteliales del túbulo renal, fibroblastos y neuronas, que hacen parte de algunos de los tejidos comprometidos en estos síndromes. Se presume que una interrupción a largo plazo de la señalización ciliar podría ser un factor de oncogénesis (Satir & Christensen, 2008).
De acuerdo con Montjean, et al., (2014) los niveles de acumulación de fosfatidilinositol 4,5-bifosfato en fibroblastos, son similares en los pacientes estudiados con síndrome de Lowe y Dent 2. Es importante destacar que en dicho estudio, se evaluó el gen INPP5B, que presenta redundancia funcional con el gen OCRL1. Sin embargo, se concluyó que dicho gen no está implicado en ninguna de estas enfermedades, puesto que, los niveles de expresión del mismo no variaron al comparar pacientes sanos con pacientes con dichos síndromes.
La mutación c.523del (Arg175Glyfs*10), está relacionada con la aparición de un codón de parada prematuro y la consecuente eliminación de 718 aminoácidos del extremo C-terminal en la proteína. Esta sección comprende los dominios de la familia endonucleasa/ exonucleasa /fosfatasa y el activador de GTPasas Rho, Rac y Cdc42 (Li, et al., 2016). En este caso, los productos sintetizados son proteínas acortadas con limitaciones funcionales y probablemente son degradadas. En algunos casos pueden verse afectados los procesos de remodelación de la membrana (Madhivanan, Mukherjee & Aguilar, 2012).
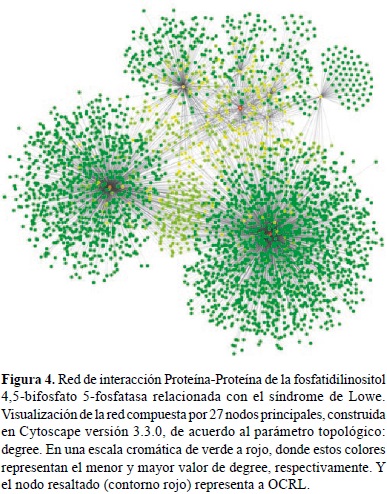
Construcción y análisis de la red de interacción proteína - proteína (IPP). Con base en la información extraída de la base de datos BioGRID se construyó una red de interacción de proteínas, relacionada con el síndrome de Lowe (Figura 4), en la cual OCRL representa el nodo principal, dado que esta directamente implicado con esta enfermedad. Los nodos adyacentes, representan las proteínas que tienen interacción directa con OCRL, y sus interacciones con otras proteínas.
La Figura 4, fue construida utilizando como atributo el parámetro topológico: Degree, que hace referencia al número de enlaces o interacciones que presenta cada nodo. De acuerdo a los valores de este parámetro en la red, podría decirse que la red presenta un modelo de libre escala, debido a que gran parte de los nodos de la red, poseen muy pocas interacciones (nodos de color verde) y pocos nodos presentan un gran número de interacciones (nodos color naranja/rojo). En este caso los nodos ELAVL1 y NXF1, tienen un valor mayor de Degree respecto a los demás. Por su parte, el nodo principal, OCRL, tiene un valor medio para este parámetro (color amarillo), esto se debe a que posee varias interacciones pero frente a los nodos ELAVL1 y NXF1 este número de enlaces es menor, sin embargo este hecho no limita las interacciones de OCRL, puesto que la mayoría de sus enlaces están relacionados directamente con la enfermedad.
Así mismo, se construyó una red de interacción proteína-proteína como se observa en la Figura 5. Donde el tamaño de los nodos y el color de los mismos representan la influencia de cada nodo en las rutas metabólicas afectadas por esta enfermedad.
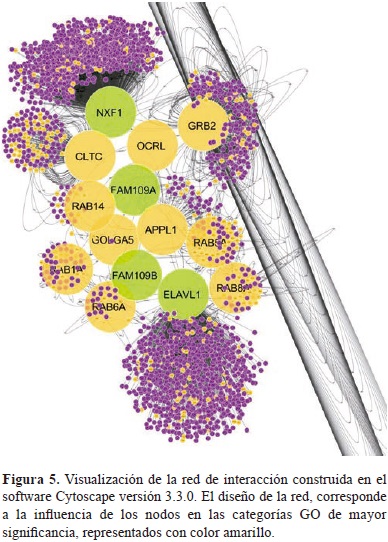
Esta escala se estableció a partir de un análisis funcional realizado con el plug-in GOlorize, el cual arrojó las categorías GO de mayor significancia y los genes vinculados a las mismas, tales como, rutas de señalización mediadas por proteínas G, actividad de GTPasas y trasporte de moléculas mediado por vesículas asociadas al aparato de Golgi, relacionadas en la Tabla 3.
En la Tabla 3 se muestran las diferentes categorías GO agrupadas de la mayor a la menor significancia, principalmente de los archivos ontológicos, procesos biológicos y componentes celulares, así como su p-val luego de la corrección de Bonferroni Family-Wise, para aumentar la confiabilidad de los datos obtenidos y evitar una sobreestimación en los mismos, además de los nodos de la red que poseen influencia sobre las mismas.
GOlorize es un plug-in diseñado para el software Cytoscape que en base a las anotaciones de Gene Ontology (GO) o información adicional dirige el proceso de diseño de gráficos de redes y destaca la función biológica de los nodos. Se utiliza en conjunto con BinGO, una herramienta eficaz para encontrar las categorías GO excesivamente representadas, ya que proporciona una red en la cual el grado de significancia de las categorías se describe cualitativamente.
En cuanto a la topología de la red, en la Tabla 4 se relacionan los valores de los parámetros topológicos para la red. El parámetro caminos mínimos, hace referencia a la distancia mínima entre un grupo de nodos frente al resto de la red, el valor medio de los caminos mínimos permite tener una idea de la cercanía entre los nodos. El mayor de los caminos mínimos representa el diámetro de la red, en este caso, el diámetro tienen un valor de 5, que corresponde a la distancia entre los dos nodos más alejados de la red. Por lo tanto, se podría afirmar que hay un alto grado de conectividad entre los nodos, puesto que entre menor sea el diámetro mayor será el grado de conectividad entre los mismos.

Por su parte, el coeficiente de clustering o transitividad cuantifica la probabilidad de que los nodos adyacentes a un nodo estén conectados. En una red o grafo, la transitividad de un nodo es 1 si todos sus nodos vecinos están conectados entre sí y 0 si no hay ninguna conexión entre ellos. Para el caso de la red, el coeficiente de transitividad es de 0,033. Esto demuestra la versatilidad de la proteína y la baja dependencia que se presenta entre los procesos celulares en los que OCRL está implicado. Es decir, si una ruta metabólica se ve afectada por una mutación en OCRL1, probablemente las demás rutas no se verán afectadas (Dent 2); salvo que la mutación comprenda variaciones en distintos dominios de la proteína y de esta manera afecte a más de una de sus funciones, como es el caso del fenotipo expresado en el síndrome de Lowe.
Conclusión
A partir de la red construida se logró apreciar de manera más clara las interacciones que presenta OCRL con otras proteínas. Así mismo, se determinó que los procesos celulares más afectados por este grupo de proteínas son: el transporte mediado por vesículas y la transducción de señales mediada por pequeñas GTPasas. El valor del coeficiente de transitividad (0.033) demuestra que las interacciones que presenta OCRL en dichos procesos celulares son independientes entre sí, puesto que, la proteína puede unirse a otras moléculas sin presentar impedimentos estéricos. Luego, cuando una mutación inhibe alguna función en la enzima, no necesariamente deben verse afectadas sus demás funciones. Esto explica claramente, porque no todas las mutaciones reportadas para este gen causan el síndrome de Lowe y por el contrario en algunos casos pueden dar origen a la enfermedad de Dent 2.
Conflicto de intereses
Los autores manifiestan que no tienen conflicto de intereses.
Referencias
Astle, M.V., Horan, K.A., Ooms, L.M., & Mitchell, C.A. (2007). The inositol polyphosphate 5-phosphatases: traffic controllers, waistline watchers and tumour suppressors? Biochem. Soc. Symp. 161-181. [ Links ]
Attree, O., Olivos, I.M., Okabe, I., Bailey, L.C., Nelson, D.L., Lewis, R.A., McInnes, R.R., and Nussbaum, R.L. (1992). The Lowe's oculocerebrorenal syndrome gene encodes a protein highly homologous to inositol polyphosphate-5-phosphatase. Nature 358: 239-242. [ Links ]
Avendaño L., Aljama P., Arias M., Caramelo C., De lo Ríos J., Lamas S. (2009). Nefrología Clinica 3ra Ed. Editorial Medica Panamericana. Madrid. 638 pp. [ Links ]
Avery G., Fletcher MA., MacDonald MG. (2001). Neonatología: Fisiopatología y manejo del recién nacido 5 ED. Editorial Medica Panamericana. Madrid. 994 pp. [ Links ]
Bökenkamp A., Böckenhauer D., Cheong H., Hoppe B., Tasic V., Unwin R., Ludwig M. (2009). Dent-2 Disease: A Mild Variant of Lowe Syndrome. The Journal of Pediatrics. Vol 155. 94-99 pp. [ Links ]
Casillas N., Villar V., Lizárraga A. (2016). Síndrome de Lowe: reporte de caso. Revista Mexicana de Oftalmología. [ Links ]
Charnas LR., Bernardini I., Rader D., Hoeg J., Gahl WA. (1991). Clinical and laboratory findings in the oculocerebrorenal syndrome of Lowe, with special reference to growth and renal function. N Engl J Med. 324: 1318-1325. [ Links ]
Chatr-Aryamontri A, Breitkreutz BJ, Oughtred R, Boucher L, Heinicke S, Chen D, Stark C, Breitkreutz A, Kolas N, O'Donnell L, Reguly T, Nixon J, Ramage L, Winter A, Sellam A, Chang C, Hirschman J, Theesfeld C, Rust J, Livstone MS, Dolinski K, Tyers M. (2014). The BioGRID interaction database: 2015 update. Nucleic Acids Research. [ Links ]
Chen, S., Zhang, X., Chen, L., Tian, Q., & Jiang, W. (2014). Analysis of OCRL gene mutation in a male infant with Lowe syndrome. Zhonghua yi xue yi chuan xue za zhi Chinese journal of medical genetics, 31 (2): 223-227. [ Links ]
Conduit A, Dyson J, Mitchell C. (2012). Inositol polyphosphate 5-phosphatases; new players in the regulation of cilia and ciliopathies. FEBS Letters. Vol 586. 2846-2857 pp. [ Links ]
Cortés V. & Villanueva C. (2015). Epidemiologia del glaucoma congenito en la Asociación para evitar la Ceguera en Mexico, revisión de 5 años. Revista Mexicana de Oftalmología. Vol 89. 150-153 pp. [ Links ]
EBSCO Industries. DynaMED Plus. [En línea]. Consultado: enero de 2016. Disponible en http://www.dynamed.com/home/. [ Links ]
Elseiver B.V. ScienceDirect. [En línea]. Consultado: enero de 2016. Disponible en http://www.sciencedirect.com/ [ Links ]
Li, F., Yue, Z., Xu, T., Chen, M., Zhong, L., Liu, T., Jing X., Deng J., Hu B., Liu Y., Wang, H. (2016). Dent Disease in Chinese Children and Findings from Heterozygous Mothers: Phenotypic Heterogeneity, Fetal Growth, and 10 Novel Mutations. The Journal of pediatrics. [ Links ]
Hoopes, R.R., Jr., Shrimpton, A.E., Knohl, S.J., Hueber, P., Hoppe, B., Matyus, J., Simckes, A., Tasic, V., Toenshoff, B., Suchy, S.F., et al. (2005). Dent Disease with mutations in OCRL1. Am. J. Hum. Genet. 76: 260-267. [ Links ]
Hou, X., Hagemann, N., Schoebel, S., Blankenfeldt, W., Goody, R. S., Erdmann, K. S., & Itzen, A. (2011). A structural basis for Lowe syndrome caused by mutations in the Rab-binding domain of OCRL1. The EMBO journal, 30 (8): 1659-1670. [ Links ]
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40 (Database issue): D109-14. [ Links ]
Krauβ, M., & Haucke, V. (2007). Phosphoinositide-metabolizing enzymes at the interface between membrane traffic and cell signalling. EMBO reports, 8 (3): 241-246.
Kornfeld M; Synder RD; MacGee J; Russell D; Appenzeller O. (1975). The Oculo-Cerebral-Renal Syndrome of Lowe Neuromuscular Components. Arch Neurol. 32 (2): 103-107. [ Links ]
Lemmon M. (2007). Pleckstrin homology (PH) domains and phosphoinositides. Biochemical Society Symposia. 81-93 pp. [ Links ]
Lin, T., Orrison, B. M., Suchy, S. F., Lewis, R. A., & Nussbaum, R. L. (1998). Mutations Are Not Uniformly Distributed throughout theOCRL1Gene in Lowe Syndrome Patients. Molecular genetics and metabolism, 64 (1): 58-61. [ Links ]
Mao, Y.; Balkin, D. M.; Zoncu, R.; Erdmann, K. S.; Tomasini, L.; Hu, F.; Jin, M. M.; Hodsdon, M. E. & De Camilli, P. (2009). A PH domain within OCRL bridges clathrin-mediated membrane trafficking to phosphoinositide metabolism. The EMBO Journal, 28 (13): 1831-1842. [ Links ]
Madhivanan, K., Mukherjee, D., & Aguilar, R. C. (2012). Lowe syndrome: between primary cilia assembly and Rac1-mediated membrane remodeling. Communicative & integrative biology, 5 (6): 641-644. [ Links ]
Madhivanan, K., & Aguilar, R. C. (2014). Ciliopathies: the trafficking connection. Traffic, 15 (10): 1031-1056. [ Links ]
Madhivanan, K., Ramadesikan, S., & Aguilar, R. C. (2015). Chapter Seven-Role of Ocrl1 in Primary Cilia Assembly. International review of cell and molecular biology, 317: 331-347. [ Links ]
McCrea, H. J., Paradise, S., Tomasini, L., Addis, M., Melis, M. A., De Matteis, M. A., & De Camilli, P. (2008). All known patient mutations in the ASH-RhoGAP domains of OCRL affect targeting and APPL1 binding. Biochemical and biophysical research communications, 369 (2): 493-499. [ Links ]
Montjean, R., Aoidi, R., Desbois, P., Rucci, J., Trichet, M., Salomon, R., Rendu J, Fauré J, Lunardi J, Gacon G, Billuart P, Dorseuil O. (2014). OCRL-mutated fibroblasts from patients with Dent-2 disease exhibit INPP5B-independent phenotypic variability relatively to Lowe syndrome cells. Human molecular genetics, ddu514. [ Links ]
Fejerman, F. Á., & Fernández Alvarez, E. (1988). Neurología pediátrica Ed.Médica Panamericana. [ Links ]
Muthappa A., Nazreen K., Al-kindi S. (2015). Duplication 1q as primary and 3q in (3;13) as secondary aberration in Fanconi anemia: Implications and literature review. Acta Haematologica Polonica. Vol 46. 368-371 pp. [ Links ]
National Center for Biotechnology Information. PubMed. [En linea]. Consultado: enero de 2016. Disponible en http://www.ncbi.nlm.nih.gov/pubmed/ [ Links ]
Núñez A., Aránguiz J., Kattan J., Escobar R. (2008). Síndrome hipotónico del recién nacido. Revista Chilena de Pediatría. Vol 79. 146-151 pp. [ Links ]
Nussbaum RL; Suchy SF. (2001). The oculocerebrorenal syndrome of Lowe (Lowe syndrome). The Metabolic and Molecular Bases of Inherited Disease Volume Ch 252. 8th edition. Edited by: Scriver CR, Beadet AL, Sly WS, Valle D. Mc-Graw Hill. New York. 6257-6266 pp. [ Links ]
Ooms, L.M., Horan, K.A., Rahman, P., Seaton, G., Gurung, R., Kethesparan, D.S., and Mitchell, C.A. (2009). The role of the inositol polyphosphate 5-phosphatases in cellular function and human disease. Biochem. J. 419: 29-49. [ Links ]
Pirruccello M & De Camilli P. (2012). Inositol 5-phosphatases: insights from the Lowe syndrome protein OCRL. Trends Biochemical Sciences, Cell press. Vol 37. 134-143 pp. [ Links ]
Sadava D. (2009). Vida: Ciencia de la Biología octava edición. Editorial Médica Panamericana. Buenos Aires. 276 pp. [ Links ]
Santucci L., Candiano G., Anglani F., Bruschi M., Tosetto E., Cremasco D., Murer L., D'Ambrosio C., Scaloni A., Petretto A., Caridi G., Rossi R., Bonanni A., Ghiggeri G. (2016). Urine proteome analysis in Dent's disease shows high selective changes potentially involved in chronic renal damage. Journal of Proteomics, 130: 26-32. [ Links ]
Satir, P., & Christensen, S. T. (2008). Structure and function of mammalian cilia. Histochemistry and cell biology, 129 (6): 687-693. [ Links ]
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research; 13 (11): 2498-504. [ Links ]
Shrimpton A.E, Hoopes, Jr. R.R, Knohl S.J, Hueber P, Reed A.A, Christie P.T, Igarashi T, Lee P, Lehman A, White C, Milford D.V, Sanchez M.R, Unwin R, Wrong O.M, Thakker R.V, Scheinman S.J. (2009). OCRL1 mutations in Dent 2 patients suggest a mechanism for phenotypic variability. Nephron Physiology, 112 (2): p27-p36. [ Links ]
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, Von Mering C. (2015). STRING v10: Protein-Portein interaction networks, integrated over the tree of life. Nucleic Acids Res. Jan;43. (Database issue): D447-52. [ Links ]
Taleisnik S. (2006). Receptores Celulares y la transducción de señales 1a ed. Encuentro Grupo Editor. Córdoba. 127-132. [ Links ]
Trésaugues, L., Silvander, C., Flodin, S., Welin, M., Nyman, T., Gräslund, S., Hammarströn, M., Berglund, H. & Nordlund, P. (2014). Structural basis for phosphoinositide substrate recognition, catalysis, and membrane interactions in human inositol polyphosphate 5-phosphatases. Structure, 22 (5): 744-755. [ Links ]
Thompson J. & Lakhani N. (2015). Cataracts. Primary Care: Clinics in Office Practice. Vol. 42. 409-423 pp. [ Links ]
Uniprot, C. (2010). Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Research 39 (Database issue): D214-D219. [ Links ]
Vicinanza, M., D'Angelo, G., Di Campli, A., & De Matteis, M. A. (2008). Function and dysfunction of the PI system in membrane trafficking. The EMBO journal, 27 (19): 2457- 2470. [ Links ]
Yepes R., Virgili C., Povedano M., Guerrero M., Villabona C. (2015). Kennedy's disease and partial androgen insensitivity síndrome. Report of 4 cases and literatura review. Endocrinología y Nutrición. Vol 62. 224-230 pp. [ Links ]

