











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroducción
Microorganismos resistentes a antibióticos. Uno de los mayores problemas actuales en salud pública es la resistencia de los microorganismos a los compuestos antimicrobianos. El uso indiscriminado de antibióticos potentes y de amplio espectro ha generado altas tasas de resistencia antimicrobiana (Schmidt, 2017). El problema se ha agravado aún más por la resistencia desarrollada por muchos patógenos frente a más de un compuesto antimicrobiano, por lo cual se les clasifica como patógenos resistentes a múltiples medicamentos (multiple drug resistance, MDR) (Mathur & Singh, 2013; Reuken, et al., 2017). Por ejemplo, el European Centre for Disease Prevention and Control (ECDC) ha informado que en Europa cada año mueren 25.000 personas debido a infecciones causadas por bacterias multirresistentes, que ocasionan costos adicionales de atención médica y pérdidas de productividad de al menos 1.500 millones de euros cada año (Li & Webster, 2018).
La Organización Mundial de la Salud (OMS) ha clasificado los patógenos de interés prioritario para el desarrollo de nuevos antibióticos (World Health Organization, WHO, 2017) de la siguiente manera: prioridad 1, crítica: Acinetobacter baumannii resistente a los carbapenémicos; Pseudomonas aeruginosa resistente a los carbapenémicos; Enterobacteriaceae resistentes a los carbapenémicos y productoras de betalactamasa de espectro extendido (ESBL); prioridad 2, elevada: Enterococcus faecium resistente a la vancomicina; Staphylococcus aureus resistente a la metici-lina con sensibilidad intermedia y resistencia a la vanco-micina; Helicobacter pylori resistente a la claritromicina; Campylobacter spp. resistentes a las fluoroquinolonas; Salmonellae resistentes a las fluoroquinolonas; Neisseria gonorrhoeae resistente a la cefalosporina y a las fluoroquinolonas, y prioridad 3, media: Streptococcus pneumoniae sin sensibilidad a la penicilina; Haemophilus influenzae resistente a la ampicilina y Shigella spp. resistentes a las fluoroquinolonas. En este contexto, las bacterias Gram negativas E. coli O157:H7 y P. aeruginosa, así como la Gram positiva Staphylococcus aureus resistente a la meticilina (SARM), se han convertido en patógenos de gran relevancia y han impulsado la búsqueda de nuevos tratamientos para combatir las infecciones que ocasionan (Brogan & Mossialos, 2016).
Estructura, propiedades bioquímicas y actividad de los péptidos antimicrobianos. Aunque el número y la diversidad de los péptidos antimicrobianos en la naturaleza son elevadas (véase la base de datos en http://aps.unmc.edu/AP/main.php), hay formas estructurales que la mayoría de ellos comparten (Cruz, et al., 2014; 2017; 2018; Prada, et al., 2016; Kumar, et al., 2018). En general, los péptidos antimicrobianos son relativamente cortos (12 a 30 aminoácidos), de naturaleza catiónica (cargados positivamente con un valor de pH neutro) y anfifílicos, con una proporción significativa de residuos hidrofóbicos. La naturaleza catiónica de la mayoría de ellos favorece un cierto grado de selectividad por los fosfolípidos de la membrana microbiana y presenta una carga generalmente catiónica (+2 a +9) y un promedio de 40 a 50 % de residuos hidrofóbicos. Estas propiedades son importantes en su mecanismo de acción antimicrobiano (Lohner, 2001).
Inicialmente, las actividades líticas de los péptidos antimicrobianos se atribuyeron a las estructuras helicoidales (Jenssen, et al., 2006). Sin embargo, se ha sugerido que tanto la estructura helicoidal como la conformación flexible son importantes para mejorar la potencia y la selectividad de los péptidos antimicrobianos no solo frente a las células bacterianas, sino también a las eucariotas (por ejemplo, para su actividad citotóxica en células plasmodiales o cancerosas). Esta flexibilidad estructural en las estructuras helicoidales de péptidos se ha asociado con la introducción cuidadosa de residuos de prolina en la secuencia del péptido antimicrobiano (Vermeer, et al., 2012).
A pesar de sus propiedades físicas similares, las homologías de secuencia de los péptidos antimicrobianos son muy limitadas y exhiben un amplio conjunto de estructuras secundarias que permiten clasificarlos a partir de sus estructuras tridimensionales (Jenssen, et al., 2006). La clasificación de los péptidos antimicrobianos es difícil debido a su gran diversidad. Según la composición de aminoácidos de los péptidos, y el tamaño y las estructuras que los conforman, los péptidos antimicrobianos pueden dividirse en varias categorías: a) péptidos con estructuras de hélice alfa, como la catelicidina humana; b) péptidos con estructuras de hoja beta estabilizadas por puentes disulfuro, como las defensinas humanas; c) péptidos con estructuras extendidas, como la indolicidina, y d) péptidos ricos en glicina, como el Pg-AMP1, y péptidos con estructuras de bucle, como las defensinas cíclicas (Li, et al., 2012).
Los mecanismos de actividad antimicrobiana de los péptidos antimicrobianos varían desde la permeabilización de la membrana hasta las acciones sobre un conjunto de moléculas intracelulares diana que incluyen las actividades inmunomoduladoras. Los péptidos pueden destruir la estructura de la membrana bacteriana produciendo la lisis celular o mediante la interacción entre péptido y membrana pueden conducir a la formación de poros transitorios y transportar el péptido dentro de la célula, poniéndolo en contacto con dianas intracelulares donde se pueden enlazar al ADN, el ARN y las proteínas, y provocar procesos como la inhibición de la síntesis de la pared celular o la síntesis de proteínas; además, pueden interferir en la citocinesis bacteriana por filamentación celular in vitro o in vivo (Teixeira, et al., 2012; Li, et al., 2017).
Diseño, síntesis y caracterización de péptidos antibacterianos. Aunque los péptidos antimicrobianos se han considerado fármacos antibióticos prometedores, son sensibles al efecto de las condiciones ambientales (pH y fuerza iónica) y a la acción de las proteasas, lo que podría limitar su aplicación terapéutica (Malik, et al, 2016). Además, son potencialmente citotóxicos para las células humanas, lo que en algunos casos ocasiona alergia, y su producción a escala industrial puede ser muy costosa (Pfalzgraff, et al., 2018).
Se ha propuesto una variedad de estrategias para mejorar la estabilidad de los péptidos frente a la degradación proteolítica. Entre ellas, se han ensayado la modificación química de extremos terminales de péptidos (Danial, et al., 2012); la alteración de sus estructuras secundarias flexibles (Rozek, et al., 2003); la estabilización de las estructuras secundarias (Houston, et al., 1998); el desarrollo de péptidos análogos mediante la sustitución con aminoácidos no naturales (por ejemplo, usando el enantiómero D) (Lee, et al., 2011; Cruz, et al., 2018); el aumento del contenido alfa helicoidal en la estructura de péptidos mediante modificaciones covalentes (Uteng, et al., 2003); la modificación de la propiedad hidrófoba, la longitud y la carga neta (Rydlo, et al., 2006); el acortamiento de péptidos antimicrobianos naturales (Lee, et al., 2011), y el diseño in silico de péptidos antimicrobianos utilizando los algoritmos genéticos (Prada, et al., 2016). Además, han surgido estrategias de diseño racional basadas en el alineamiento de secuencias de los péptidos antimicrobianos en una plantilla. En estudios previos (Yang, et al., 2017) se ha modificado un único aminoácido en la secuencia, con el objetivo de determinar las posiciones y los residuos que son importantes para la actividad antimicrobiana. Algunos péptidos, como la cecropina, la manganina, la temporina, la protegrina y la lactoferricina, se han utilizado como plantillas de péptidos antimicrobianos (Merlino, et al., 2017).
Una vez que se ha diseñado in silico la nueva secuencia de un péptido antimicrobiano, se debe hacer su síntesis. Una de las metodologías más utilizadas en la síntesis de péptidos en fase sólida (SPFS) es la basada en el 9-fluorenilmetilcarboxil (Fmoc), debido a sus altos rendimientos (Jaradat, 2018). Este mecanismo consiste en la elongación de una cadena peptídica anclada a un soporte solido mediante la adición sucesiva de aminoácidos que se unen a través de un enlace amida (enlace peptídico) entre el grupo carboxilo del aminoácido entrante y el grupo amino del aminoácido previamente ligado al soporte hasta obtener la secuencia del péptido deseado. Los dos métodos de síntesis peptídica habitualmente usados son el Fmoc y el basado en el tercbutiloxicarbonilo (t-Boc), los cuales difieren en los reactivos y condiciones empleadas para llevarlos a cabo (Jaradat, 2018).
En cuanto a la purificación del péptido antimicrobiano sintetizado, el grado de pureza se determina usualmente mediante cromatografía liquida de alta resolución en fase reversa (reversed phase high-performance liquid chromatography, RP-HPLC) (Aguilar, 2004). Para la determinación de la masa molecular del péptido, se utilizan la espectrometría de masas (mass spectometry, MS), dada su sensibilidad, velocidad y alto grado de especificidad molecular (Il'ina, et al., 2011), y el dicroísmo circular (DC), técnica alternativa de espectroscopía de absorción electrónica, que permite determinar la estructura secundaria del péptido. Hoy existen métodos de deconvolución que utilizan bases de datos y permiten establecer una estructura muy acertada (Bakshi, et al., 2014). En este contexto, en el presente estudio se describen el diseño, la síntesis, la purificación, la caracterización bioquímica y la evaluación antimicrobiana de péptidos antimicrobianos contra E. coli, S. aureus y P. aeruginosa resistentes a antibióticos.
Materiales y métodos
Materiales. Los aminoácidos L obtenidos con el Fmoc se adquirieron en IRIS Biotech GmbH (Germany). La resina de rink-amida, y los reactivos y disolventes orgánicos para la síntesis de péptidos se obtuvieron en Merck Novabiochem® (Germany). El 2,2,2-trifluoroetanol (TFE) y el ácido α-ciano-4-hidroxicinámico (ACC) se adquirieron de Sigma-Aldrich (St. Louis, USA). Se usó Agua Milli-Q® con una resistividad de 18,3 MΩ α 25 °C para la preparación de todas las soluciones.
Microorganismos. Las tres cepas bacterianas, E. coli O157: H7, P. aeruginosa y SARM, fueron donadas por la Escuela de Microbiología de la Pontificia Universidad Javeriana. Las líneas celulares de tipo epitelial de carcinoma de pulmón humano A549 y la HepG2 fueron donadas por el Laboratorio de oxidación de cultivos celulares y biológicos de la Universidad Federal de Rio de Janeiro (Brasil).
Los medios de cultivo Müller Hilton (MH), la infusión de cerebro y corazón (BHI) y el Luria Bertani (LB) se adquirieron de Oxoid (Basingstoke, Inglaterra). El medio M199 se adquirió de Gibco (Invitrogen, CA). Los reactivos de lisostafina, bromuro de 3-(4,5-dimetiltiazol-2-ilo)-2,5- difeniltetrazol (MTT), dimetilsulfóxido (DMSO) y Tritón® X-100 se obtuvieron en Sigma-Aldrich (St. Louis, USA). Para obtener las diferentes concentraciones de péptido, se preparó una solución madre con una concentración de 1 mg de péptido por ml de agua o 1 mM de tampón de fosfato de sodio con pH de 7.2.
Diseño bioinformático de péptidos. El diseño de nuevos péptidos con potencial antimicrobiano se hizo mediante el uso de máquinas de soporte vectorial (support vector machines, SVM) utilizando el modelo denominado relación cuantitativa entre estructura y actividad (quantitative structure-activity relationship, QSAR), para el reconocimiento de patrones y la creación de algoritmos que permitieran detectar la actividad antibacteriana mediante una estrategia de optimización basada en algoritmos genéticos, con la cual se hace una búsqueda de péptidos antimicrobianos con rangos establecidos de carga, carácter hidrófobo, punto isoeléctrico e índice de inestabilidad, y que se denominó DEPRAMS (programa desarrollado en nuestro grupo GIBIM) (Trindade, et al., 2014). Las secuencias peptídicas se alinearon con la herramienta BLASTP y el método de alineamiento múltiple de secuencia Clustal W, con el fin de obtener residuos conservados en los nuevos péptidos diseñados, y cambiar aminoácidos de la secuencia del péptido para dar origen a péptidos análogos también con propiedades antimicrobianas. Como moldes se usaron diferentes proteínas homólogas, todas publicadas en la base de datos del National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/blast.cgi). Se utilizó la base de datos denominada CAMP para determinar teóricamente los valores de las propiedades fisicoquímicas de los péptidos diseñados (propiedad hidrófoba, índice alifático, carga neta, punto isoeléctrico, peso molecular y porcentaje teórico de ser un péptido antimicrobiano según dichas características) (Conlon, et al., 2011).
Síntesis de péptidos antimicrobianos. Los péptidos se obtuvieron mediante la síntesis química en fase sólida (solid-phase peptide synthesis, SPPS), empleando la estrategia Fmoc en bolsas de polipropileno. Para sintetizar el péptido desde su extremo C-terminal se empleó una resina de rink-amida 4MBHA (capacidad: 0,63 meq/gramo). Las bolsas cargadas con 100 mg de resina se marcaron y sellaron. Los aminoácidos correspondientes en cada secuencia se acoplaron con N,N-diisopropiletilamina (DIPEA) como agente activador y O-(benzotriazol-1-il)-N,N,N,N-tetrametiluronio tetrafluoroborato (TBTU) como agente anti-racémico (7 equivalentes de DIPEA por 5 equivalentes de TBTU) para acoples de menos de 3 horas, y N,N-diciclohexilcarbodiimida (DCC) con el 1-hidroxibenzotriazol (HOBt) para los acoples de 12 horas (Strateva & Yordanov, 2009). El indicador azul de bromofenol (1 % p/v en dimetilformida, DMF) se añadió a la solución al finalizar cada etapa de acoplamiento para verificar de forma cualitativa la eficiencia de los acoples en la reacción. La desprotección del grupo Fmoc se llevó a cabo por piperidina al 20 % v/v en DMF mediante dos lavados de 10 minutos cada uno (Babu & Gopi, 1998). Finalizada la elongación del péptido, se hizo el clivaje con el objetivo de desprender el péptido de la resina usada como soporte de la síntesis. Los péptidos se clivaron de la resina con una mezcla de ácido trifluoroacético, agua y trihidroxopropilsilano (TFA/H2O/TIS, 95:2,5:2,5); posteriormente, se dejaron en agitación durante 3 horas, después se filtró la resina y, por último, se precipitó la solución filtrada con 5 mL de éter frío. La aparición de un sólido blanco lechoso correspondiente al péptido sintetizado se centrifugó a .3500 rpm y a 4 °C durante 15 minutos (Broekman, et al., 2011). Los péptidos se purificaron mediante RP-HPLC (Jasco Corporation, Tokio, Japón) usando una columna Vydac® C18 y una mezcla de H2O con TFA al 0,1 % (v/v) y acetonitrilo (ACN) que contenía TFA al 0,1 % (v/v) como fase móvil. Para la elución de los péptidos se utilizó el siguiente programa de gradiente: 30 minutos con 5 a 70 % de la segunda mezcla con flujo de 1 mL/min, y detección en una longitud de onda (λ) de 220 nm (Jofré, et al., 2011).
Caracterización de los péptidos sintéticos. La masa molar de los péptidos purificados se determinó mediante espectrometría de masas MALDI-TOF como se ha descrito previamente (Fields & Noble, 2009), usando un espectrómetro de masas Bruker UltrafleXtrem™ en modo reflectrón y utilizando el ácido α-ciano-4-hidroxicinámico (HCCA) como matriz. Posteriormente, se determinó la actividad antimicrobiana de los péptidos contra E. coli O157:H7, P. aeruginosa y SARM mediante microdilución en caldo.
La estructura secundaria de los péptidos se determinó mediante dicroísmo circular (DC) (Aguilar, 2004) utilizando un espectrómetro CD (J-815, Jasco Corporation, Japón). Los péptidos se ajustaron a una concentración final de 200 µM en una solución al 30 % (v/v) de 2,2,2-trifluoroetanol (TFE), preparando 200 µL de volumen final que contenían 60 microlitros de 2,2,2 TFE, agua y una muestra de péptido. La concentración del péptido se determinó con base en su peso molecular y su coeficiente de extinción molar. En el coeficiente de extinción molar calculado se tuvo en cuenta el número de residuos de triptófano y tirosina. Se calculó el respectivo volumen de péptido para obtener una concentración final de 200 µM. La elipticidad molar [9] de cada péptido se calculó mediante la siguiente ecuación: [θ]=3298.2 ∆ε, donde s es el coeficiente de extinción molar.
La concentración de los péptidos se determinó midiendo la absorbancia de las soluciones a 220 nm con la ecuación de Lambert-Beer y teniendo en cuenta la absorción del enlace peptídico.
Determinación de la actividad antimicrobiana in vitro de los péptidos antimicrobianos libres. Para determinar la actividad antimicrobiana de los péptidos diseñados y sus análogos contra E. coli O157:H7, P. aeruginosa y SARM, en la estimación de los valores de la CIM se utilizó el método de microdilución en placas de 96 pozos según los protocolos descritos por varios autores (Abercrombie, et al., 2015; Cruz, et al., 2017).
Para determinar la CMI, la absorbancia de las micro-placas se midió en un espectrofotómetro de microplaca Elisa (BIO-RAD, Imark™) a 595 nm, 37 °C y 200 rpm. Inicialmente, se preparó un pre-inóculo tomando 2 a 3 colonias de E. coli O157:H7, P. aeruginosa y SARM previamente sembradas en agar BHI para SARM y LB para P. aerugionsa y E. coli, e incubando a 37 °C durante 24 horas. Las microplacas contienen diluciones seriadas de los péptidos antimicrobianos libres (0,5, 5, 10, 25, 50, 75 y 100 µM). La CMI se definió como la menor concentración de péptido antimicrobiano que produjera una inhibición del crecimiento de la bacteria. Los valores de CMI50 y CMI99 corresponden a las dosis que inhiben el 50 y el 99 % del crecimiento, respectivamente. La CMB se define como la concentración en la cual hubo 100 % del crecimiento bacteriano en comparación con el control positivo utilizado (la bacteria en crecimiento en el medio de cultivo apropiado) (Yoon, et al., 2014; Cruz, et al., 2014).
Evaluación del efecto de las sales en la actividad antibacteriana de los péptidos antimicrobianos. Para evaluar la sensibilidad salina de los péptidos más activos, se preparó una solución madre de 200 µM para cada sal (NaCl, KCl y MgCl2) y se esterilizó para tomar el volumen respectivo de cada una y obtener concentraciones finales de 150 mM de NaCl, 4.5 mM de KCl y 1 mM de MgCl2 en el pocillo de la microplaca, con el fin de evaluar la actividad antimicrobiana según el procedimiento ya descrito.
Determinación de la actividad hemolítica de los péptidos La actividad hemolítica de los péptidos GIBIM-P1 a GIBIM-P6 y sus análogos se determinó midiendo en eritrocitos de carnero la hemólisis inducida por estos compuestos, como se ha hecho en estudios previos (Lozano, et al., 2010), y utilizando concentraciones del péptido antimicrobiano de 5 a 100 ¡iM. Como control positivo se utilizó una suspensión que contenía los eritrocitos y Tritón® X-100 al 1 % (v/v). Esta combinación produce el 100 % de hemólisis. El porcentaje de hemólisis se calculó mediante la siguiente ecuación:
Hemolisis (%) = (As-Ao)/(A100-A0) x100 %, donde As es la absorbancia de la muestra, A100 es la absorbancia de los eritrocitos sometidos a lisis completa en 0,1 % de Tritón® X-100 y A0 es la absorbancia en ausencia completa de hemólisis.
Determinación de la citotoxicidad de los péptidos antimicrobianos in vitro. La citotoxicidad de los péptidos con mayor actividad antimicrobiana se determinó in vitro con el método publicado por Lozano, et al. (2010) y Chaudhary (2016). Para el ensayo se cultivaron las líneas celulares de carcinoma de pulmón humano A549 y HepG2 en medio Eagle modificado (EMEM) complementado con suero fetal bovino (SFB) al 10 % (p/v), en el cual se colocaron 7.500 células por pocillo en microplacas de 96 pocillos con un volumen final de 0,2 mL; a continuación, se incubaron en una atmósfera humidificada de CO2 al 5 % (v/v), a 37 °C durante 24 horas. La viabilidad celular se determinó midiendo la absorbancia a 550 nm en un lector de microplacas (Multiskan de Thermo Scientific™). El control positivo de la lisis celular se hizo con Triton X-100 al 0,1 % (v/v), el cual ocasionó una lisis del 100 %.
Análisis estadístico. Los datos experimentales se representan como media ± desviación estándar. Las diferencias entre los grupos se examinaron mediante un análisis unidireccional de la varianza utilizando el sistema SPSS 19.0 (IBM Corp., Armonk, NY, EE.UU.). Se consideró que un valor de p<0,05 indicaba una diferencia estadísticamente significativa.
Resultados y discusión
Diseño de nuevos péptidos antimicrobianos. Los péptidos y sus análogos se diseñaron in silico como péptidos antibacterianos de tipo hélice alfa y se visualizaron con el programa PEP-FOLD 3.5. En la figura 1 se presentan los seis péptidos denominados GIBIM-P1 a GIBIM-P6, los cuales se usaron posteriormente como plantilla para el desarrollo de 12 nuevos análogos. La estructura secundaria se simuló en línea usando el servidor de predicción PEP-FOLD 3.5 (http://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::PEP-FOLD3).

Figura 1 Simulación de la estructura secundaria de los péptidos diseñados usados como secuencia original. La construcción de la estructura secundaria in silico (teórica) se realizó en la base de datos pep-fold 3.5 teniendo en cuenta la estructura cristalográfica de las proteínas que contiene la base de datos, mediante homología de secuencia, utilizando el algoritmo DEPRAMS.
Las secuencias de los péptidos inicialmente diseñados y de sus análogos se presentan en la Tabla 1. Las modificaciones en la secuencia de los péptidos GIBIM-P2 a GIBIM-P5, usadas para diseñar los análogos peptídicos, buscaban aumentar la carga neta positiva, la naturaleza hidrófoba y el porcentaje de probabilidad de ser un péptido antimicrobiano, propiedades fisicoquímicas directamente relacionadas con la actividad antimicrobiana (Il'ina, et al., 2011; Bakshi, et al., 2014). Por esta razón, el diseño de los péptidos análogos GIBIM-P3A6K, GIBIM-P5T2K y GIBIM-P5S9K, y los residuos de alanina (A), treonina (T) y serina (S) se reemplazaron por lisina (K) para aumentar el carácter catiónico de los péptidos (Teixeira, et al., 2012; Sengupta, et al., 2008; Vila-Farrés, et al., 2012).
Tabla 1 Propiedades fisicoquímicas de los péptidos diseñados y sus análogos
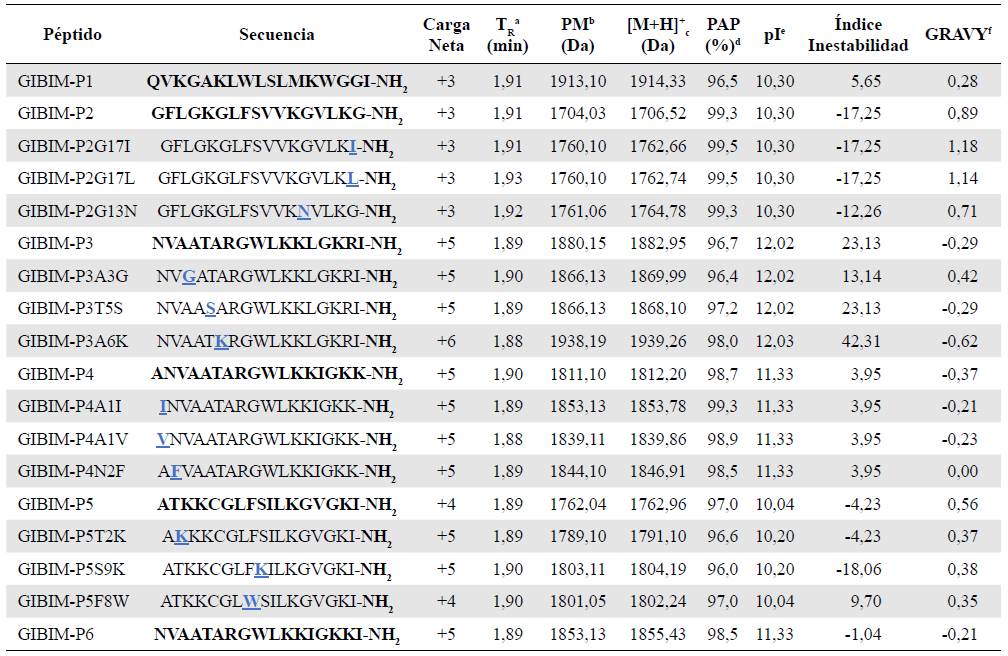
aTr: tiempo de retención en min; bPeso Molecular; cRelación masa/carga; "Probabilidad de ser péptido antimicrobiano; ePunto Isoeléctrico; Promedio del índice de Hidropatía; b Peso molecular; c Relación masa/carga; d Probabilidad de ser péptido antimicrobiano; e Punto isoeléctrico; f Promedio del índice de hidropatía
En el diseño de los péptidos análogos GIBIM-P2G17I, GIBIM-P2G17L, GIBIM-P4A1I, GIBIM-P4A1V y GIBIM-P4N2F, los aminoácidos que aparecen subrayados se cambiaron por isoleucina (I), leucina (L), valina (V) y fenilalanina (F), respectivamente, para aumentar su carácter hidrófobo (Yu, et al., 2017). Por último, en los péptidos antimicrobianos GIBIM-P3A3G, GIBIM-P3T5S y GIBIM-P5F8W, los aminoácidos subrayados se sustituyeron por asparagina (N), glicina (G), serina (S) y triptófano (W), respectivamente, para aumentar la probabilidad de ser péptido antimicrobiano (Yang, et al., 2017).
La naturaleza catiónica de los péptidos antimicrobianos representa el principal factor de contacto con la superficie bacteriana de carga opuesta a través de interacciones electrostáticas para facilitar su acción (Yeaman & Yount, 2003). Además, la presencia de aminoácidos específicos en la posición precisa de la cadena peptídica es igualmente crucial para la expresión de la actividad antimicrobiana. Esto sucede probablemente porque los aminoácidos poseen una gran variedad de propiedades fisicoquímicas y cada uno tiene un único grupo funcional de cadena lateral. Además, la asignación de aminoácidos en la posición precisa de la cadena peptídica aseguraría la integridad estructural y la estabilidad de los péptidos antimicrobianos de forma conservada para garantizar la interacción específica con la diana. En cuanto a su característica de hidrófobos, esta les permite a los péptidos penetrar en las células e inducir la lisis de las membranas (Aoki & Ueda, 2013). Además, el aumento de la naturaleza hidrófoba de la secuencia de aminoácidos de los péptidos antimicrobianos se correlaciona con su baja selectividad y toxicidad en las células mamíferas. Asimismo, la estructura anfipática de los péptidos antimicrobianos facilita su interacción con la membrana celular y su posterior inserción en la matriz de los fosfolípidos (Tossi, et al., 2000).
Por lo anterior, en el diseño de los péptidos análogos GIBIM-P3A6K, GIBIM- P5T2K y GIBIM-P5S9K, los residuos de alanina (A), treonina (T) y serina (S), respectivamente, se reemplazaron por lisina (K) para aumentar el carácter catiónico de los péptidos. En el diseño de los péptidos análogos GIBIM-P2G17I, GIBIM-P2G17L, GIBIM-P4A1I, GIBIM-P4A1V y GIBIM-P4N2F, los aminoácidos subrayados se cambiaron por isoleucina (I), leucina (L), valina (V) y fenilalanina (F), respectivamente, con el fin de aumentar su carácter hidrófobo. Por último, en los péptidos GIBIM- P2G13N, GIBIM-P3A3G, GIBIM-P3T5S y GIBIM-P5F8W, los aminoácidos subrayados se sustituyeron por asparagina (N), glicina (G), serina (S) y triptófano (W), respectivamente, con el fin de aumentar la probabilidad probabilidad de que constituyeran péptidos antimicrobianos.
Los 18 péptidos antimicrobianos diseñados tuvieron una probabilidad de serlo mayor al 96 % y un índice de ines-tabilidad menor de 40, lo que garantiza que tendrían un tiempo de vida media in vivo mayor a 16 horas (Tripathi, et al., 2017), excepto en el caso del péptido GIBIM-P3A6K. Las características fisicoquímicas de los péptidos se presentan en la Tabla 1. Los péptidos GIBIM-P6, GIBIM-P3, GIBIM-P4 y los análogos de los dos últimos obtuvieron la mayor carga neta positiva (entre 5 y 6), seguidos del péptido GIBIM-P5 y sus análogos (carga neta entre 4 y 5). No obstante, los péptidos más hidrofóbicos fueron GIBIM-P2G17I y GIBIM-P2G17L, con un índice de hidropatía de 1,18 y 1,14, respectivamente. Las modificaciones en las secuencias peptídicas originales no afectaron la estructura secundaria de hélice alfa de los 12 análogos, la cual conservó la misma estructura anfipática.
Síntesis y caracterización de los péptidos. Los 18 pép-tidos alcanzaron un grado de pureza aproximado de 95 a 98 % (Tabla 1). Cuando se determinó la pureza del péptido sintetizado, se midió la absorbancia en tres longitudes de onda: 210, 220 y 280 nm. La primera no se tomó en cuenta porque absorbió la mayoría de los enlaces presentes en la muestra. La segunda correspondía a la absorción del enlace peptídico y la tercera a los grupos de anillos aromáticos que se encuentran en las cadenas laterales de los aminoácidos fenilalalanina (F), triptófano (W) y tirosina (Y). Si los péptidos poseen alguno de estos aminoácidos en la secuencia, pueden cuantificarse a 280 nm. Sin embargo, si este grupo de aminoácidos no está presente en la secuencia, se cuantifica la concentración del péptido a 220 nm teniendo en cuenta la absorción del enlace peptídico.
En todos los casos fue posible confirmar la secuencia primaria de los péptidos GIBIM-P1 a GIBIM-P6 y sus análogos mediante el análisis de masas MALDI-TOF. Las masas moleculares teórica y experimental de cada péptido se presentan en la tabla 1. Los resultados obtenidos mediante el análisis con esta técnica sugieren que los péptidos se sintetizaron satisfactoriamente, ya que los datos experimentales de unidades de masa (m/z) para cada uno concordaron con sus respectivas masas moleculares obtenidas teóricamente.
Además, se determinó la proyección de la rueda helicoidal de Schiffer-Edmundson para cada péptido (no se muestran los datos). Los diagramas revelaron una conformación de carácter anfipático para los péptidos, mostrando que todos los residuos hidrófilos de estos compuestos están situados en un lado de la hélice, en tanto que los residuos hidrófobos están en el otro lado de esta.
En cuanto a la estructura secundaria de los péptidos sintéticos y análogos, se determinó el espectro de dicroísmo circular en condiciones simuladas de membrana en presencia de TFE/H2O a una concentración de 30 % (v/v). Los péptidos adoptaron una estructura de hélice alfa y exhibieron un mínimo de dos bandas negativas a 205 y 215 nm, lo que indica que adoptan una estructura de hélice alfa bien definida (Figura 2). En la Tabla 2 se observa que todos los péptidos tuvieron un porcentaje helicoidal mayor al 2,4 % (v/v), siendo el análogo GIBIM-P2G17L el de menor estructura helicoidal, en tanto que el péptido GIBIM-P3 exhibió la mayor estructura helicoidal, la cual se tomó como referencia (100 %). Estos resultados indicaron que la cara hidrófoba no disruptiva a lo largo de la cara no polar del péptido estabiliza la estructura helicoidal.
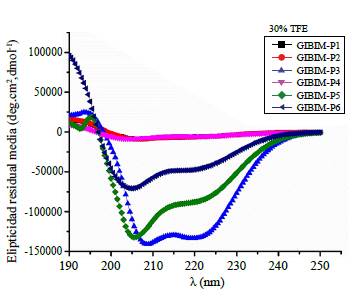
Figura 2 Espectros de DC de los péptidos diseñados. Se promediaron los valores de tres scans por muestra, y las concentraciones de péptido se fijaron a 150 |iM. Los péptidos análogos adoptaron una estructura alfa-helicoidal similar (datos no mostrados). Todos los péptidos tuvieron un porcentaje de estructura helicoidal mayor al 2.4%, siendo el análogo GIBIM-P2G17L con el menor contenido
Tabla 2 Datos del DC dicroísmo circular de los péptidos
| Peptido | [θ] 222* | % Helice |
|---|---|---|
| GIBIM-P1 | -5584,01 | 4,3 |
| GIBIM-P2 | -6078,55 | 4,6 |
| GIBIM-P2G17I | -11985,60 | 9,2 |
| GIBIM-P2G17L | -3162,54 | 2,4 |
| GIBIM-P2G13N | -5916,88 | 4,5 |
| GIBIM-P3 | -130804,00 | 100,0 |
| GIBIM-P3A3G | -85721,20 | 65,5 |
| GIBIM-P3T5S | -5493,67 | 4,2 |
| GIBIM-P3A6K | -8515,40 | 6,5 |
| GIBIM-P4 | -5253,31 | 4,0 |
| GIBIM-P4A1I | -4241,47 | 3,2 |
| GIBIM-P4A1V | -82558,70 | 63,1 |
| GIBIM-P4N2F | -40880,20 | 31,3 |
| GIBIM-P5 | -85015,50 | 65,0 |
| GIBIM-P5T2K | -34259,10 | 26,2 |
| GIBIM-P5S9K | -23084,00 | 17,6 |
| GIBIM-P5F8W | -19221,30 | 14,7 |
| GIBIM-P6 | -44706,50 | 34,2 |
Evaluación de la actividad antibacteriana de los péptidos ntimicrobianos. Los péptidos catiónicos diseñados inhibieron el crecimiento de las bacterias estudiadas de forma dependiente de la dosis de la concentración (Tabla 3). En general, al menos 11 de los 18 péptidos evaluados mostraron ser activos frente a las cepas patógenas en una concentración igual o inferior a 100 uM. En todos los ensayos de actividad antibacteriana se usó la ofloxacina como control positivo, ya que es un antibiótico de amplio espectro contra bacterias Gram positivas y negativas.
Tabla 3 Actividad antibacteriana de los péptidos GIBIM-P1 a GIBIM-P6 y análogos
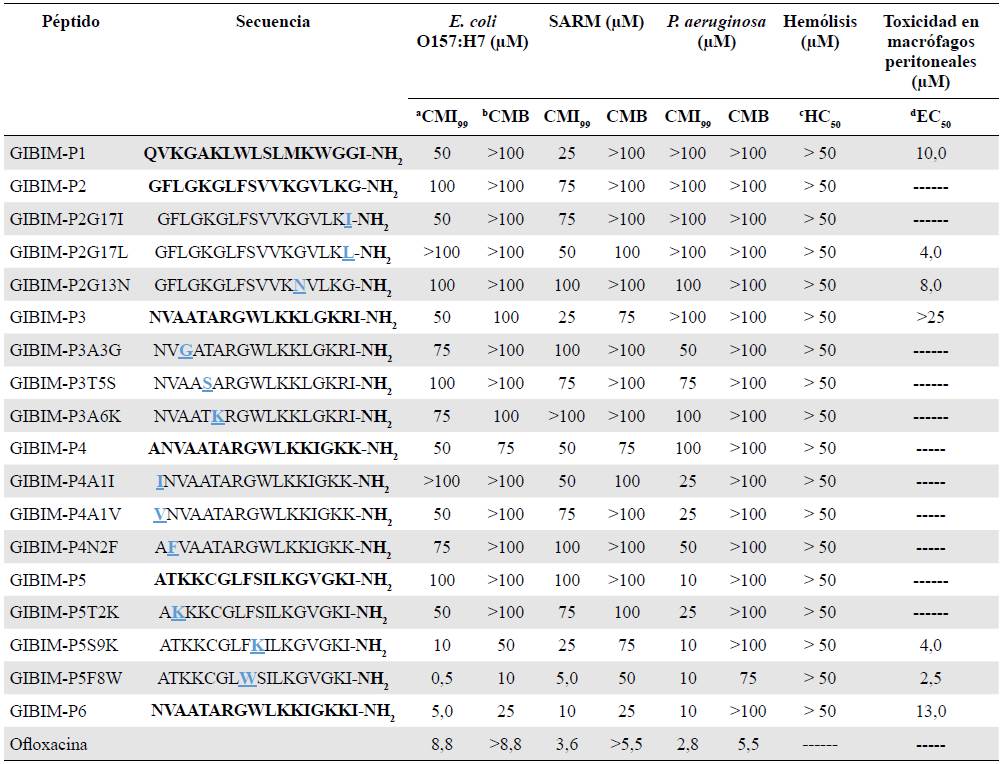
Los datos que se muestran son el promedio de tres experimentos independientes realizados por triplicado.
"Concentración Mínima Inhibitoria; bConcentración Mínima Bactericida; cHC50 Concentración hemolítica al 50%; dEC50 concentración efectiva al 50%
El péptido con mayor actividad antibacteriana frente a las cepas evaluadas fue el GIBIM-P6, con una CMI99 de 5,0 µM frente a E. coli O157: H7, y de 10 µM contra SARM y P. aeruginosa. En segundo lugar, los tres péptidos antimicrobianos GIBIM-P4, GIBIM-P3 y GIBIM-P5 presentaron una CMI99 de 50 µM frente a E. coli O157:H7, de 25 µM frente a SARM y de 10 µM frente a P. aeruginosa. No obstante, los análogos de péptidos antimicrobianos incrementaron su actividad antibacteriana. Por ejemplo, los péptidos antimicrobianos análogos GIBIM-P5F8W y GIBIM-P5S9K exhibieron, respectivamente, una CMI de 5,0 y de 25 µM frente a S. aureus; de 50 y 100 µM frente a E. coli O157: H7 y, más interesante aún, de 10 µM frente a P. aeruginosa. Estos análogos se diseñaron a partir de la sustitución de un aminoácido de la secuencia del péptido GIBIM-P5, con lo cual se logró aumentar su carácter hidrófobo (cambiando F por W) y su carga (cambiando S por K). Por el contrario, algunas modificaciones fueron desfavorables y disminuyeron la actividad de los análogos con respecto al péptido de referencia, lo cual indica que las sustituciones en la región C-terminal no producen efecto sobre el carácter hidrófobo y, por lo tanto, no afectan la actividad antimicrobiana. Sin embargo, otros estudios han demostrado que este tipo de sustituciones pueden afectar la estabilidad del péptido antimicrobiano (Berthold, et al., 2013).
Por otro lado, la actividad de los péptidos antimicrobianos GIBIM-P1, GIBIM-P3 y GIBIM-P4 presentaron importantes valores de CMI de 50 µM y 25 µM frente a E. coli O157:H7 y SARM, respectivamente. Cabe resaltar que todos los péptidos antimicrobianos diseñados y sintetizados se encontraban en forma de amidas en la región C-terminal, con el fin de neutralizar la carga negativa creada por el grupo carboxílico C-terminal. Esta modificación se añade para evitar la degradación enzimática, favorecer la imitación de proteínas nativas y, en algunos casos, evitar la unión del extremo C-terminal de los péptidos con iones de hidrógeno u otros grupos reactivos que podrían ocasionar interferencia en los ensayos (Kim, et al., 2011).
Los péptidos antimicrobianos son moléculas muy específicas, por este motivo pueden interactuar de forma diferente con las bacterias patógenas y afectar su actividad. Así, si se compara la actividad de los péptidos antimicrobianos (Tabla 4) frente a las dos cepas Gram negativas P. aeruginosa y E. coli O157:H7, es posible observar que presentan un mayor efecto sobre esta última cepa. En la Tabla 3 se observa una CMI99 de 10 uM frente a E. coli O157:H7 cuando se cultiva en presencia del péptido control (es decir, en ausencia de sales), en tanto que en presencia de Mg+2, la CMI99 aumenta a 12,5 µM. Este efecto podría deberse a que la permeabilidad de membrana externa de esta bacteria es extremadamente escasa, haciendo que las bombas de eflujo constituyan un mecanismo de defensa contra estas moléculas (Andersson, et al., 2016).
Tabla 4 Actividad antibacteriana de los péptidos GIBIM-P5S9K y GIBIM-P5F8W en presencia de NaCl 150 mM, KCl 4.5 mM y MgCl2 1 mM, respectivamente, expresada en CMI99 (µM)
| Péptidos | E. coli O157:H7 | SARM | P. aeruginosa | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control | Na+1 | K+1 | Mg+2 | Control | Na+1 | K+1 | Mg+2 | Control | Na+1 | K+1 | Mg+2 | |||
| GIBIM-P5S9K | 10 | 10 | 10 | 12,5 | 25 | 25 | 25 | 25 | 10 | 10 | 10 | 10 | ||
| GIBIM-P5F8W | 0,5 | 0,5 | 0,5 | 5,0 | 5,0 | 5,0 | 5,0 | 5,0 | 10 | 10 | 10 | 10 | ||
Los péptidos antimicrobianos GIBIM-P1, GIBIM-P3 y GIBIM-P5S9K presentaron una elevada actividad antibacteriana contra SARM, alcanzando valores comparables a los de otros péptidos cíclicos anfifílicos y sus análogos, los cuales han sido diseñados contra patógenos multirresistentes, como es el caso del péptido cíclico [R4W4], el péptido antimicrobiano más potente contra SARM descrito hasta la fecha, con una CMI99 de 2,67 µg/mL (Oh, et al., 2014). Asimismo, se demostró el potencial de la estrategia propuesta para diseñar y optimizar péptidos: el GIBIM-P5S9K aumentó diez veces la actividad antimicrobiana comparado con el péptido original GIBIM-P5 contra E. coli y cuatro veces contra SARM, y fue más eficiente que el GIBIM-P5T2K frente a estas bacterias. Las modificaciones en el GIBIM-P5T2K y el GIBIM-P5S9K se hicieron en la cara hidrófoba de la hélice alfa. En ambos casos el reemplazo fue similar: Thr y Ser se cambiaron por Lys.
Los resultados sugieren que el aumento de la actividad antimicrobiana no se debe exclusivamente al aumento de la carga positiva global del péptido, sino a la distribución de cargas positivas a lo largo de su estructura. De hecho, el GIBIM-P3A6K (con la carga neta positiva más alta, +6) no aumentó su actividad en comparación con el GIBIM-P3, a pesar de que el efecto electrostático entre el péptido y la membrana bacteriana es impulsado por las interacciones iónicas entre los grupos catiónicos del péptido y la cabeza aniónica de los fosfolípidos, como ya se ha evidenciado (Shai, et al., 2002). Esto es muy importante si se tiene en cuenta que tanto las bacterias Gram positivas como las Gram negativas desarrollan diferentes estrategias para generar resistencia a los péptidos antimicrobianos, aunque las estructuras celulares de su envoltura son distintas (Joo, et al., 2016).
Los péptidos antimicrobianos GIBIM-P1, GIBIM-P3 y GIBIM-P4N2F presentaron menor actividad que otros péptidos de composición similar (Zhu, et al., 2014), probablemente porque la ubicación de los residuos de Trp en la secuencia no permite la interacción п-catión, con una estabilización concomitante de los anillos indoles aromáticos y una mayor actividad antibacteriana (Epand, et al., 1999; Schibli, et al, 2002; Chan, et al, 2006). En el caso del GIBIM-P5F8W, su porcentaje de homología es del 92 % en 11 aminoácidos con el péptido antimicrobiano anfibio escuelentin (Tennessen & Blouin, 2010). Este péptido potenció su actividad 220 veces comparado con el péptido inicial GIBIM-P5 frente a E. coli O157:H7 (MIC99=0.5 µM) y SARM (MIC99=5.0 µM), respectivamente.
La sustitución del aminoácido de Trp en una de las regiones hidrófobas tiene un papel importante en el aumento de la actividad antimicrobiana y ha sido estudiada por otros autores (Chen, et al., 2005; Lee, et al., 2013). Este resultado indica la importancia especial del aminoácido de Trp en la región media de la secuencia peptídica, y demuestra que este residuo no solo se divide más favorablemente en la interfase de membrana, sino que también es más hidrófobo y tiene una mayor afinidad por fases hidrofóbicas voluminosas que otros residuos aromáticos (Zhu, et al., 2014). Otro péptido antimicrobiano muy activo fue el GIBIM-P6, el cual exhibió actividad frente a E. coli O15.H7, con una MIC99 de 5,0 µM. Asimismo, frente a SARM y P. aeruginosa registró una MIC99 de 10,0 µM. Sin embargo, este compuesto se oxida fácilmente a temperatura ambiente y su manipulación a nivel experimental fue muy complicada.
En este contexto, al combinar un método que utiliza un modelo computacional para correlacionar las propiedades fisicoquímicas de un péptido y un diseño racional que identifica los aminoácidos conservados, se obtuvieron seis péptidos con la mejor actividad antimicrobiana, es decir, cuatro péptidos diseñados (GIBIM-P3, GIBIM-P4, GIBIM-P5 y GIBIM-P6) y dos péptidos análogos (GIBIM-P5S9K y GIBIM-P5F8W), siendo estos dos últimos muy eficaces y adecuados como candidatos a ser agentes antimicrobianos. Sin embargo, algunos de los nuevos péptidos producidos en este estudio tienen actividad antibacteriana comparable o superior a péptidos como el cecropin B, el cecropin P1 y el hepcidin (Sengupta, et al., 2008; Vila-Farrés, et al., 2012).
Además, el GIBIM-P5S9K registró un 92 % de homología en diez aminoácidos con el péptido antimicrobiano anfibio esculentina. Esto significa que, de alguna manera, el algoritmo puede imitar la evolución natural en algunos péptidos antimicrobianos.
Por otra parte, uno de los múltiples obstáculos para el uso de péptidos antimicrobianos en la clínica es la disminución de la actividad antimicrobiana en presencia de sales fisiológicas, debido a que los cationes compiten por los sitios de unión a la membrana bacteriana con carga negativa (Zhu, et al., 2014; Kandasamy & Larson, 2006). Por lo tanto, se determinó el efecto de algunas sales en concentraciones fisiológicas en la actividad antimicrobiana de los péptidos GIBIM-P5S9K y GIBIM- P5F8W. Como se observa en la Tabla 3, los péptidos presentaron el mismo efecto antibacteriano independientemente de la sal utilizada. Sin embargo, los péptidos GIBIM-P5S9K y GIBIM-P5F8W disminuyeron su actividad frente a la cepa bacteriana de E. coli O157:H7 en presencia de la sal con mayor carga catiónica (MgCl2, mM). En estudios previos (Kandasamy & Larson, 2006) se ha evidenciado que la unión de péptido y lípido es más fuerte con concentraciones más bajas de sal. Los péptidos alteran los lípidos, pero este efecto disminuye a medida que aumenta la concentración de sal. Además, es probable que los residuos catiónicos de los péptidos compitan con los cationes monovalentes y divalentes de las sales en los mismos sitios de unión de la región de la cabeza hidrofóbica del lípido y por ello disminuyan su actividad.
Toxicidad in vitro de los péptidos activos. Para ser útiles en la aplicación terapéutica, los péptidos antimicrobianos deben ser selectivos frente a las células procariotas, por tal razón, se evaluó la actividad hemolítica y la toxicidad en líneas celulares. Los resultados de la hemólisis indicaron que ninguno de estos péptidos presentó una actividad hemolítica superior al 40 % con una concentración de los péptidos de 50 uM (Figura 3), pues para ser hemolíticos debían producir una hemólisis mayor al 50 % con concetraciones de 25 µM con respecto al control (Fernandez-Reyes, et al., 2010).
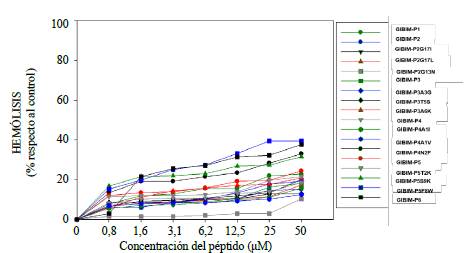
Figura 3 Actividad hemolítica de los PAMs diseñados y sus análogos frente a eritrocitos de carnero. Se utilizaron concentraciones del pétpido comprendidas entre 5 y 100 µM, usando como control positivo una suspensión que contenía los eritrocitos y Tritón X-100 al 1%(v/v).
Por otro lado, se evaluó el efecto citotóxico de los péptidos antimicrobianos en la línea celular A549 de tejido pulmonar, la cual se incubó en presencia de diferentes concentraciones de los péptidos con mayor actividad antibacteriana (Figura 4A). Estos péptidos no registraron ninguna citotoxicidad detectable, incluso en una concentración de 100 µM, con excepción del péptido GIBIM-P5F8W, el cual presentó un porcentaje de células viables por debajo del 50 % (36,8 %) con 100 µM (lo que es 200 a 10 veces mayor que la CMI de las cepas evaluadas). Además, la viabilidad de esta 9l9ínea celular incubada con el péptido GIBIM-P5S9K a 100 µM (que es cuatro veces mayor que la CMI99), fue del 50 % en comparación con las células no tratadas. Los péptidos se consideran citotóxicos cuando, a una concentración cuatro veces por encima de la MIC , presentan un porcentaje de células viables menor al 50 % (Nakhjavani, et al., 2014).
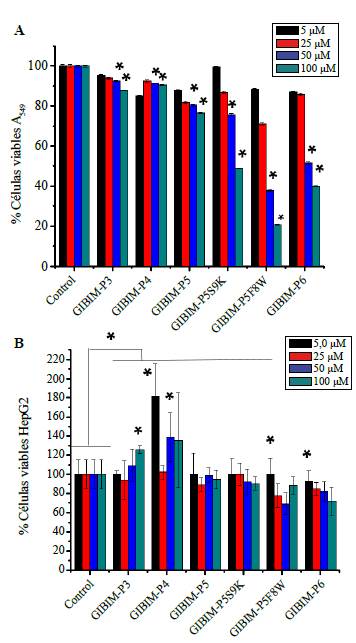
Figura 4 Citotoxicidad de los péptidos más activos en líneas celulares. A) Línea celular A549 y B) línea celular HepG2. Los datos que se muestran resultan del promedio de tres experimentos independientes realizados por triplicado. *P<0.05.
Además, en la evaluación de la citotoxicidad de los péptidos antimicrobianos más activos frente a las células HepG2 de carcinoma humano, se observó que los péptidos GIBIM-P3 y GIBIM-P4 promovieron el crecimiento celular en algunas de las concentraciones evaluadas (Figura 4B). Estos resultados son similares a los obtenidos por Yang, et al. (2017), en los cuales el péptido GRP (VPLPAGGGTVLTKMYPR GNHWAVGHLM-NH) sirvió como un mitógeno para las células HepG2, dando lugar a una mejor proliferación, una reducción de la apoptosis y la aceleración de la progresión del ciclo celular. Por su parte, los péptidos GIBIM-P5, GIBIM-P5S9K, GIBIM-P5F8W y GIBIM-P6 presentaron un porcentaje de viabilidad celular de 94,5, 90,7, 88,6 y 72,3 %, respectivamente. Los ensayos citotóxicos in vitro demostraron que los péptidos evaluados no presentaron efectos sobre la viabilidad de los eritrocitos y las líneas celulares A549 y HepG2.
Los resultados obtenidos sugieren que los péptidos más activos, GIBIM-P5S9Ky GIBIM-P5F8W, pueden servir como agentes terapéuticos frente a infecciones causadas por E. coli 0157:H7, SARM y P. aeruginosa, ya que presentan una elevada actividad antimicrobiana y un bajo efecto citotóxico.
Conclusiones
En el presente estudio se demostró que es posible diseñar y sintetizar péptidos con secuencias cortas (17 Aa) y con actividad antimicrobiana contra bacterias patógenas resistentes a antibióticos utlizando el análisis bioinformático racional. Asimismo, mediente técnicas de diseño racional es posible mejorar las propiedades bioquímicas de los péptidos, incrementar su actividad antibacteriana y mitigar su toxicidad. Por ejemplo, el aumento de las cargas positivas de los péptidos, así como el aumento de su naturaleza hidrófoba, incidieron positivamente en la actividad antibacteriana contra E. coli, P. aeruginosa y SARM. La actividad antibacteriana de los péptidos antimicrobianos sintetizados frente a E. coli fue mayor que la actividad antibacteriana frente a SARM y P. aeruginosa. Las CMI contra E. coli fueron de 0,5 µM, 5,0 y 10 µM con los péptidos GIBIM-P5F8W, GIBIM-P6 y GIBIM-P5S9K, respectivamente, siendo valores menores a los publicados en los artículos consultados. La toxicidad de los péptidos en los eritrocitos produjo un porcentaje de hemólisis menoral 40 %a 50 µM, y en las líneas celulares A549 y HepG2 el único compuesto que presentó toxicidad fue el GIBIM-P5F8W, el cual dejó un 36 % de células viables con 100 µM en la línea A549. Además, los péptidos de GIBIM- P3 y GIBIM- P4 indujeron el crecimiento de las células HepG2 en ciertas concentraciones. Por último, se encontró que los péptidos antimicrobianos más potentes frente a las bacterias patógenas fueron el GIBIM-P5S9K y el GIBIM-P5F8W. Estos adoptan una estructura helicoidal alfa en ambientes diseñados para imitar membranas celulares y son capaces de permear la membrana de bacterias patógenas de una manera dependiente de la dosis. Además, pueden considerase como compuestos selectivos debido a su baja toxicidad en las células eucariotas.

