



![Electronic and electrochemical studies of 2,2-bis[5'-(2'-carbonylfuranyl)]propane](/img/en/prev.gif)







 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introduction
The control of the selectivity remains as one of the main concerns in the synthetic organic chemistry (Balcells, et al., 2016; Ward, 1999). Particularly, selective reduction of C=C double bonds in the presence of carbonyl and carboxyl functionalities (in α,β-unsaturated systems), is frequently a challenging aspect in synthetic organic strategies. For that, several approaches have been reported in that direction with varied grades of success (Cornubert & Eggert, 1954; Cornubert & Thomas, 1954; Hammond, et al., 2009; Inoue, et al., 2002; Chikashita, et al., 1987; Felpin & Fouquet, 2010; Gutierrez, et al., 2010; Zheng, et al., 2015; Keess & Oestreich, 2017; Mori, et al., 2006; Lipshutz, et al., 1998; Wang, et al., 2016). In a classical procedure described by Cornubert et al., the selective reduction of the C=C double bond in the α,β-unsaturated ketone (chalcone) 1a was performed by means of a catalytic hydrogenation process in EtOH mediated by Raney-Nickel poisoned with halo-alkanes (R-X). Nevertheless, appreciable formation of the double-reduction side-product (i.e. saturated alcohol 3a) also accompanied this process, Scheme 1(Cornubert & Eggert, 1954; Cornubert & Thomas, 1954). When reaction was performed in absence of halo-alkanes, the double-reduction side-product 3a was obtained exclusively (Tucker, 1950).
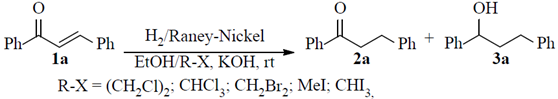
Scheme 1 Attempts to selective reduction of the C=C double bond in chalcone 1a by catalytic hydrogenation using Raney-Nickel poisoned with halo-alkanes (R-X).
A modification to the above approach was performed by Hammond, et al., replacing the reducing couple H2/Raney-Nickel by ammonium formate/Pd/C, affording the expected product 2a in better yield (Hammond, et al., 2009). Alternatively, NaBH4-based chemical reductions have also been performed but in most cases the carbonyl or both carbonyl and C=C double bond functionalities resulted in reduction (Zeynizadeh & Shirini, 2003; Aramini, et al., 2000; Mohamadi, et al., 2013).
As part of our current program on the synthetic utilization of chalcones and their analogues for the synthesis of diverse heterocyclic compounds (Insuasty, et al., 2017; Ramírez-Prada, et al., 2017; Montoya, et al., 2016; Insuasty, et al., 2016), herein, we wish to report two complementary approaches for the preparation of diverse α,β-reduced derivatives of synthetic interest mediated by the selective (catalytic and chemical) reduction of their α,β-C=C double bonds starting from their corresponding α,β-unsaturated precursors.
Materials and Methods
Melting points were determined on a Stuart SMP10 melting point apparatus and are uncorrected. FTIR spectra were recorded on a Shimadzu FTIR 8400 ATR technique. 1H and 13C NMR spectra were recorded on a Bruker Avance 400 spectrophotometer operating at 400 MHz and 100 MHz respectively, using CDCl3 and DMSO-d6, as solvents and tetramethylsilane (TMS) as internal standard. Mass spectra were run on a SHIMADZU-GCMS-QP-2010 Plus spectrometer (equipped with a direct inlet probe) operating at 70 eV. The starting indole was purchased from Merck (analytical grade reagent), and was used without further purification. The starting N-alkylindoles (i.e. 1-methyl, 1-butyl and 1-benzyl) were synthesized by N-alkylation reaction using iodomethane, 1-bromobutane and benzyl chloride, following the procedures described by (Kong, et al., 2006). Synthetic details for the new and some representative known compounds are described as follow:
Selective reduction of the α,β-unsaturated C=C double bonds in chalcones 1 by catalytic hydrogenation
A mixture of chalcone 1 (1 gram), ethanol (5-10, mL), chloroform or DCM (0.5 mL) and Raney-Nickel (0.3 g) was stirred at room temperature under hydrogen pressure (20 psi) in a Parr apparatus for 15-20 min. The solid formed was completely dissolved by adding the adequate volume of ethyl acetate, the catalyst was removed by filtration and product recovered after removing the solvent mixtures under reduced pressure. No further purification was required to afford hydrogenated ketones 2.
Synthesis of 1-methyl-1H-indole
In a 15 mL round-bottom flask, NaH (2 mmol) was added to a cold (0 °C) stirred solution of indole (1.0 mmol) in THF (5 mL). After 25 min, iodomethane (1.4 mmol) was added at 0 °C and the mixture was stirred at room temperature for 3 h. The solution was then quenched with a saturated solution of ammonium chloride (4 mL), and extracted with ethyl acetate (2 x 5 mL). The organic layers were combined, washed with brine, dried over anhydrous MgSO4 and concentrated under reduced pressure. The resulting oil was purified by flash chromatography (eluent: hexane/dichloromethane 40%), to provide the corresponding product as a yellow oil in 98% yield. FTIR (ATR): v = 3010 (C-H), 2946 (C-H), 2929 (C-H), 2900 (C-H), 2842 (C-H), 1570 (C=C), 1550 (C=C) cm-1.
General procedure for the synthesis of 1-benzyl- and 1-butyl-1H-indoles
Triturated KOH (1.5 g, 27.3 mmol, 3.2 equiv) was added to a stirred solution of indole (1.0 g, 8.53 mmol, 1 equiv) in acetone (15 mL). After 20 min of stirring benzyl chloride (1.73 g, 13.6 mmol, 1.6 equiv) was added. The solution was stirred at room temperature until the indol was no longer detected by TLC. The insoluble materials were removed by filtration, and the acetone was concentrated under reduced pressure. The resulting oil was purified by flash chromatography (eluent: hexane/dichloromethane 1:1). The same procedure was followed by using 1-bromobutane instead benzyl chloride.
1-Benzyl-1H-indole. Yellow oil, 86 % yield. FTIR (ATR): v = 3005 (C-H), 2940 (CH), 2889 (C-H), 1585 (C=C), 1560 (C=C) cm-1. 1H NMR (400 MHz, CDCl3): δ = 5.40 (s, 2H, CH2Ph), 6.68 (d, J = 3.0 Hz, 1H), 7.05 - 7.53 (m, 9H), 7.79 (d, J = 7.7 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 50.2 (CH2Ph), 101.8, 109.8, 119.6, 121.1, 121.8, 126.9, 127.7, 128.3, 128.8 (Cq), 128.9, 136.4 (Cq), 137.7 (Cq) ppm.
1-Butyl-1H-indole. Yellow oil, 82 % yield. FTIR (ATR): v = 3007 (C-H), 2956 (C-H), 2929 (C-H), 2872 (C-H), 1609 (C=C), 1483, 1463, 1456, 1315, 1242, 1199, 1012, 734, 713 cm-1. 1H NMR (400 MHz, CDCl3): δ = 1.11 (t, J = 8.4 Hz, 3H, CH3), 1.51 (m, J = 15.1, 7.4 Hz, 2H, CH2), 1.97 (m, J = 7.3 Hz, 2H, CH2), 4.25 (t, J = 7.1 Hz, 2H, CH2), 6.68 (s, 1H, =C-H), 7.24 (d, J = 2.8 Hz, 1H, ArH), 7.29 (t, J = 6.7 Hz, 1H, ArH), 7.40 (t, J = 6.0 Hz, 1H, ArH), 7.52 (d, J = 8.2 Hz, 1H, ArH), 7.83 (d, J = 7.8 Hz, 1H, =C-H) ppm. 13C NMR (100 MHz, CDCl3): δ = 13.9 (CH3), 20.4 (CH2), 32.5 (CH2), 101.0, 46.2 (CH2), 109.6, 119.3, 121.1, 121.4, 127.9, 128.7 (Cq), 136.1 (Cq) ppm.
General procedure for the synthesis of indole-oxo-propanenitriles
These compounds were obtained by adapting procedures described previously by (Fadda, et al., 2014; Cai, et al., 2018; Chen, et al., 2013). In a 25 mL round-bottom flask coupled with a condenser and stir bar, a mixture of the corresponding indole (1.0 mmol), cyanoacetic acid (1.2 mmol) and acetic anhydride (2-3 mL) was stirred under heating in an oil bath at 105 °C for 1.5 h. During this time, the initial yellow solution turned dark-red and a precipitate was observed. After complete consumption of the starting indole (monitored by TLC), the solution was cooled to 0 °C overnight. The precipitate was filtrated and washed with methanol (2 x 1.0 mL). No further purification was required in any case.
3-(1H-Indol-3-yl)-3-oxopropanenitrile. This compound was obtained according to the above procedure from indole as beige solid, 79% yield, mp 243 °C. FTIR (KBr): v = 3225 (N-H), 2253 (CN), 1643 (C=O), 1581 (C=C) cm-1. 1H NMR (400 MHz, DMSO-d6): 5 = 4.47 (s, 2H, CH2), 7.19-7.27 (m, 2H), 7.47-7.52 (m, 1H), 8.10-8.16 (m, 1H), 8.36 (s, 1H), 12.14 (s, 1H, NH) ppm. MS (70 eV): m/z (%) = 184 (23) [M]+ •, 144 (100), 116 (27), 89 (26).
3-(1-Methyl-1H-indol-3-yl)-3-oxopropanenitrile. This compound was obtained according to the above procedure from 1-methyl-1H-indole as a brown solid, 73% yield, mp 153 °C. FTIR (ATR): v = 3110 (C-H), 2250 (CN), 1643 (C=O), 1581 (C=C) cm1. 1H NMR (400 MHz, CDCl3): δ = 3.88 (s, 2H, CH2), 3.90 (s, 3H, CH3), 7.31-7.48 (m, 3H), 7.81 (s, 1H), 8.27-8.37 (m, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 29.6 (CH2), 33.9 (CH3), 110.1, 114.2 (Cq), 114.9 (CN), 122.3, 123.5, 124.2, 126.1 (Cq), 136.2, 137.5 (Cq), 180.5 (C=O).
3-(1-Benzyl-1H-indol-3-yl)-3-oxopropanenitrile. This compound was obtained accord- ing to the above procedure from 1-benzyl-1H-indole as a beige solid, 72% yield, mp 124 °C. IR (ATR): ν = 3105 (C-H), 2252 (CN), 1662 (C=O), 1602 (C=C) cm-1. 1H NMR (400 MHz, CDCl3): δ = 3.90 (s, 2H, CH2CN), 5.41 (s, 2H, CH2Ph), 7.21 (dd, J = 7.5, 2.1 Hz, 2H), 7.30-7.40 (m, 6H), 7.85 (s, 1H), 8.37 (dd, J = 8.4, 1.7 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 29.8 (CH2), 51.1 (CH2Ph), 110.6, 114.7 (Cq), 114.8 (CN), 122.5, 123.6, 124.4, 126.3 (Cq), 127.2, 128.5, 129.2, 135.1 (Cq), 135.2, 137.1 (Cq), 180.6 (C=O). Anal. Calcd. For C18H14N2O (274.32): C, 78.81; H, 5.14; N, 10.21. Found: C, 78.93; H, 5.35; N, 9.94.
3-(1-Butyl-1H-indol-3-yl)-3-oxopropanenitrile. This compound was obtained according to the above procedure from 1-butyl-1H-indole as a yellow solid, 81% yield, mp 108 °C. FTIR (ATR): v = 2249 (C=N), 1641 (C=O) cm-1. 1H NMR (400 MHz, CDCl3): δ = 0.97 (t, J= 7.4 Hz, 3H), 1.38 (m, J = 7.4 Hz, 2H), 1.88 (m, J = 7.4 Hz, 2H), 3.88 (s, 2H), 4.18 (t, J = 7.2 Hz, 2H), 7.42 - 7.29 (m, 3H), 7.82 (s, 1H), 8.34 - 8.27 (m, 1H) ppm. 13C NMR (400 MHz, CDCl3): δ = 13.6 (CH2), 20.1 (CH2), 29.7 (CH2), 31.8 (CH2), 47.2 (CH2), 110.3, 114.2, 115.0 (Cq), 122.5, 123.4, 124.1, 126.3 (Cq), 135.1, 136.9 (Cq), 180.5 (C=O) ppm.
General procedure for the synthesis of the α,β-unsaturated α-cyanoketones 4
In a round-bottom flask coupled with a condenser and stir bar, a mixture of the oxo-propanenitrile derivative (1.0 mmol), the corresponding aldehyde (1.2 mmol), triethylamine (TEA) (0.2 mL) and ethanol (4 mL) was stirred during 2 h at reflux, observing the formation of a yellow precipitate (corresponding to the α-cyanoketone 4). When the starting oxo-propanenitrile derivative was completely consumed, the solution was cooled to room temperature and the precipitate was filtrated and washed with methanol to provide the corresponding α-cyanoketone 4. No further purification was required.
(E)-2-Benzoyl-3-(2-bromophenyl)acrylonitrile (4a). This compound was obtained according to the above procedure from benzoyl acetonitrile and 2-bromobenzaldehyde as an orange solid, 52% yield, mp 126 °C. FTIR (ATR): v = 3015 (C-H), 2225 (CN), 1664 (C=O), 1251, 1024 cm-1. 1H NMR (400 MHz, CDCl3): δ = 7.39 (t, J = 7.7 Hz, 1H), 7.49 (t, J = 7.7 Hz, 1H), 7.54 (t, J = 7.6 Hz, 2H), 7.65 (t, J = 7.4 Hz, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.92 (d, J = 8.0 Hz, 2H), 8.35 (s, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ =113.7 (Cq), 115.8 (CN), 126.5 (Cq), 128.2, 128.9, 129.6, 130.2, 132.2 (Cq), 133.7, 133.8, 135.5 (Cq), 154.1, 188.7 (C=O) ppm.
(E)-3-(2-Bromophenyl)-2-(1-methyl-1H-indole-3-carbonyl)acrylonitrile (4d). This compound was obtained according to the above procedure from 3-(1-methyl-1 H-indol-3-yl)-3-oxopropanenitrile and 2-bromobenzaldehyde as a yellow solid, 92% yield, mp 163 °C. FTIR (ATR): v = 3018 (C-H), 2212 (CN), 1635 (C=O), 1581 cm1. 1H NMR (400 MHz, CDCl3): δ = 3.95 (s, 3H), 7.35 - 7.57 (m, 5H), 7.75 (dd, J = 8.0, 1.3 Hz, 1H), 8.20 (dd, J = 7.8, 1.7 Hz, 1H), 8.35 (s, 1H), 8.44 - 8.54 (m, 1H), 8.58 (s, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 34.0 (CH3), 110.0, 113.9 (Cq), 114.1 (Cq), 117.8 (CN), 123.1, 123.5, 124.3, 126.1 (Cq), 127.4 (Cq), 128.0, 130.0, 132.8 (Cq), 132.9, 133.5, 137.2, 137.4 (Cq), 152.2, 178.6 (C=O) ppm. MS: (70 eV) m/z (%) = 366/364 (40/38) [M]+ •, 285 (100), 236 (19), 158 (66), 152 (35). Anal. Calcd. For C19H13BrN2O (365.23): C, 62.48; H, 3.59; N, 7.67. Found. C, 62.22; H, 3.56; N, 8.00.
(E)-3-(2-Bromophenyl)-2-(1H-indole-3-carbonyl)acrylonitrile (4e). This compound was obtained according to the above procedure from 3-(1H-indol-3-yl)-3-oxopropanenitrile and 2-bromobenzaldehyde as a yellow solid, 95% yield, mp 249 °C. FTIR (ATR): v = 3174 (N-H), 3012 (C-H), 2218 (CN), 1631 (C=O), 1579 cm-1. 1H NMR (400 MHz, DMSO-d6): δ = 7.26-7.35 (m, 2H) , 7.53 (t, 1H ), 7.58 (d, 1H), 7.64 (t, 1H), 7.86 (d, 1H), 8.13 (d, 1H), 8.22 (d, 1H), 8.28 (s, 1H), 8.52 (s, 1H), 12.39 (s, 1H, NH) ppm. 13C NMR (400 MHz, DMSO-d6): δ =113.1 (Cq), 113.9, 115.9 (CN), 117.0, 121.8, 122.8, 123.1 (Cq), 125.0, 126.5, 128.7(Cq), 130.6 (Cq), 133.2 (Cq), 133.7, 134.8, 136.8 (Cq), 137.2, 151.0, 180.9 (C=O) ppm. MS: (70 eV) m/z (%) = 352/350 (22/22) [M]+ •, 144 (100), 271 (41), 116 (37), 89 (62).
(E)-2-(1-Benzyl-1H-indole-3-carbonyl)-3-(2-bromophenyl)acrylonitrile (4f). This compound was obtained according to the above procedure from 3-(1-benzyl-1H-indol-3-yl)-3-oxopropanenitrile and 2-bromobenzaldehyde as a yellow solid, 92% yield, mp 180 °C. FTIR (ATR): v = 3018 (C-H), 2225 (CN), 1656 (C=O), 1517 cm1. 1H NMR (400 MHz, CDCl3): δ = 5.44 (s, 2H), 7.29 - 7.19 (m, 2H), 7.45 - 7.33 (m, 7H), 7.50 (td, J = 7.7, 1.2 Hz, 1H), 7.73 (dd, J = 8.0, 1.1 Hz, 1H), 8.19 (dd, J = 7.8, 1.6 Hz, 1H), 8.36 (s, 1H), 8.63 - 8.46 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3): 5 = 51.2 (CH2Ph), 110.5, 114.2 (Cq), 114.5 (Cq), 117,4 (CN), 123.1, 123.6, 124.4, 126.1 (Cq), 127.3, 127.6 (Cq), 128.0, 128.5, 129.2, 130.0, 132.7 (Cq), 132.9, 133.4, 135.1 (Cq), 136.6, 137.0 (Cq), 151.9, 178.3 (C=O) ppm. MS: (70 eV) m/z (%) = 442/440 (8/8) [M]+ •, 361 (12), 234 (5), 91 (100).
(E)-2-(1-Benzyl-1H-indole-3-carbonyl)-3-(2-chloroquinolin-3-yl)acrylonitrile (4g). This compound was obtained according to the above procedure from 3-(1-benzyl-1H-indol-3-yl)-3-oxopropanenitrile and 2-chloroquinoline-3-carbaldehyde as a yellow solid, 89 % yield, mp 199 C. FTIR (KBr): v = 3057, 2999, 2958 (C-H), 2245 (CN), 1649 (C=O), 1589 cm-1. 1H NMR (400 MHz, CDCl3): δ = 5.44 (s, 2H), 7.30 - 7.27 (m, 2H), 7.44 - 7.33 (m, 6H), 7.73 - 7.64 (m, 1H), 7.93 - 7.84 (m, 1H), 8.00 (d, J = 7.9 Hz, 1H), 8.09 (d, J = 8.5 Hz, 1H), 8.37 (s, 1H), 8.51 (dd, J = 8.5, 1.2 Hz, 1H), 8.63 (s, 1H), 9.03 (s, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 51.1 (CH2), 110.4, 14.2 (Cq), 114.9 (Cq), 1117.2 (Cq), 122.9, 123.6, 124.4, 124.9 (Cq), 126.3 (Cq), 127.2, 127.3 (Cq), 127.9, 128.2, 128.4, 128.6, 129.0, 132.6, 134.8 (Cq), 136.3, 136.8 (Cq), 138.9, 147.6, 147.9 (Cq), 149.6 (Cq), 178.2 (C=O) ppm. MS: (70 eV) m/z (%) = 449/447 (3/10), 234 (7), 91 (100).
(E)-3-(2-Chloroquinolin-3-yl)-2-(1-methyl-1H-indole-3-carbonyl)acrylonitrile (4h). This compound was obtained according to the above procedure from 3-(1-methyl-1H-indol-3-yl)-3-oxopropanenitrile and 2-chloroquinoline-3-carbaldehyde as a yellow solid, 70 % yield, mp 175 C. FTIR (KBr): v = 2958, 2929 (C-H), 2225 (CN), 1645 (C=O), 1589 cm-1. 1H NMR (400 MHz, CDCl3): δ = 3.92 (s, 3H), 7.39 - 7.36 (m, 3H), 7.66 (t, J = 7.5 Hz, 1H), 7.90 - 7.83 (m, 1H), 7.98 (d, J = 8.1 Hz, 1H), 8.05 (d, J = 8.4 Hz, 1H), 8.33 (s, 1H), 8.46 (dd, J = 6.7, 3.0 Hz, 1H), 8.65 (s, 1H), 9.00 (s, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 34.0, 110.0, 114.1 (Cq), 114.9 (Cq), 117.7 (Cq), 123.1, 123.6, 124.4, 125.2 (Cq), 126.5 (Cq), 127.4 (Cq), 128.1, 128.5, 128.8, 132.8, 137.2, 137.4 (Cq), 138.9, 148.1, 148.3 (Cq), 149.9 (Cq), 177.9 (C=O) ppm.
(E)-2-(1-Butyl-1H-indole-3-carbonyl)-3-(2-chloroquinolin-3-yl)acrylonitrile (4i). This compound was obtained according to the above procedure from 3-(1-buthyl-1H-indol-3-yl)-3-oxopropanenitrile and 2-chloroquinoline-3-carbaldehyde as a yellow solid, 83 % yield, mp 147 C. FTIR (KBr): v = 2951, 2929 (C-H), 2250 (CN), 1645 (C=O), 1525 (C=C) cm-1. 1H NMR (400 MHz, CDCl3): δ = 0.98 (t, J = 7.4 Hz, 3H), 1.42 (m,2H), 1.92 (m, 2H), 4.24 (t, J = 7.1 Hz, 2H), 7.47 - 7.36 (m, 3H), 7.66 (t, J = 7.6 Hz, 1H), 7.86 (t, J = 7.7 Hz, 1H), 7.99 (d, J = 8.2 Hz, 1H), 8.06 (d, J = 8.5 Hz, 1H), 8.36 (s, 1H), 8.51 - 8.44 (m, 1H), 8.65 (s, 1H), 9.02 (s, 1H) ppm. MS: (70 eV) m/z (%) = 415/413 (17/53), 378 (90), 200 (100), 144(58), 41 (33).
Methyl (E)-2-cyano-3-(4-methoxyphenyl)acrylate (12). This compound was obtained according to the above procedure from p-anisaldehyde (0.200 g, 1.47 mmol), methyl 2-cyanoacetate (0.220 g, 2.22 mmol), TEA (3 drops) under stirring at 70 °C for 2 h. The reaction produced a white solid, 95% yield, mp 64 °C. 1H NMR (400 MHz, CDCl3): δ = 3.85 (s, 3H), 3.86 (s, 3H), 7.37 (s, 4H), 7.72 (s, 1H). Other spectroscopic data for this compound were already reported in the literature. CAS 14479-58-4. https://pubchem.ncbi.nlm.nih.gov/substance/318298647#section=Top
(E)-2-Cyano-N-(2-iodophenyl)-3-phenylacrylamide (14). This compound was obtained starting from the (E)-2-cyano-3-phenylacrylic acid (0.300 g, 1.73 mmol) dissolved in 2.0 mL of thionyl chloride, the mixture was stirred at reflux for 1 h, after that, the excess of thionyl chloride was removed under reduced pressure. The acyl chloride (yellow solid) formed was dissolved in 5 mL of chloroform and 2-iodoaniline (0.380 g, 1.74 mmol) and TEA (5 drops) was added. The solution was stirred at room temperature by 2 h until the formation of a precipitate. The beige solid was filtered and dried at ambient, 62% yield, 140-142 oC. 1H NMR (400 MHz, CDCl3): δ = 6.95 (td, J = 7.7, 1.5 Hz, 1H), 7.44 (ddd, J = 8.4, 7.5, 1.4 Hz, 1H), 7.48 - 7.67 (m, 3H), 7.88 (dd, J = 8.0, 1.4 Hz, 1H), 7.99 - 8.10 (m, 2H), 8.37 (dd, J = 8.2, 1.5 Hz, 1H), 8.47 (s, 1H), 8.53 (s, 1H, NH) ppm. 13C NMR (100 MHz, CDCl3): δ = 90.5 (Cq), 104.3(Cq), 116.6 (CN), 122.1, 126.9, 129.3, 129.4, 130.9, 131.7 (Cq), 133.2, 137.6 (Cq), 139.2, 154.0, 158.5 (C=O) ppm.
General procedure for the synthesis of partially-reduced compounds 5, 7, 9, 11, 13 and 15 via selective chemical NaBH-mediated reduction of the double bond C=C in their α,β-unsaturated precursors 4, 6, 8,10,12 and 14, respectively
In an Erlenmeyer with a stir bar, precursor 4, 6, 8, 10, 12 or 14 (0.2-1.0 g, 1.0 equiv.) was dissolved in a mixture dioxane/methanol (1:4) (5-10 mL). The solution was stirred at 70 °C, then, NaBH4 (1.0 equiv.) was added portionwise until the starting material was no longer detected by TLC. The solution was cooled at room temperature and the solvent was removed under reduced pressure. Water was added to the reaction flask, and the solid formed was filtered, washed with water and with cold methanol to provide the corresponding product.
2-(2-Bromobenzyl)-3-oxo-3-phenylpropanenitrile (5a). This compound was obtained according to the above procedure from 4a (0.200 g) as a yellow oil. FTIR (ATR): v = 3111, 2245 (CN), 1695 (C=O), 1257, 1026 cm-1. 1H NMR (400 MHz, CDCl3): δ = 3.31 (dd, J = 13.8, 9.7 Hz, 1H), 3.58 (dd, J = 13.8, 5.9 Hz, 1H), 4.84 (dd, J = 9.7, 5.9 Hz, 1H), 7.21 (td, J = 7.7, 1.8 Hz, 1H), 7.77 - 7.31 (m, 6H), 8.17 - 7.97 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3): δ = 36.1 (CH2), 39.2 (CH), 111.1, 116.6, 124.2, 128.1, 128.9, 129.1, 129.6, 132.3, 133.1, 134.7, 135.2, 189.9 (C=O) ppm.
2-(2-Bromobenzyl)-3-(1-methyl-1H-indol-3-yl)-3-oxopropanenitrile (5d). This compound was obtained according to the above procedure from 4d (1.000 g) as a beige solid, mp 146 °C. FTIR (ATR): v = 3059 (C-H), 2249 (CN), 1641 (C=O), 1577 cm1. 1H NMR (400 MHz, CDCl3): δ = 3.38 (dd, J = 13.8, 9.5 Hz, 1H), 3.61 (dd, J = 13.7, 6.1 Hz, 1H), 3.89 (s, 3H), 4.53 (dd, J = 9.5, 6.1 Hz, 1H), 7.19 (td, J = 7.7, 1.7 Hz, 1H), 7.28 - 7.50 (m, 5H), 7.61 (dd, J = 8.0, 1.3 Hz, 1H), 7.89 (s, 1H), 8.43 (ddd, J = 6.8, 3.0, 1.5 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 33.9 (NCH3), 36.5 (CH2), 40.4 (CH), 110, 114.1 (Cq), 117.9 (CN), 122.7, 123.5, 124.2 (Cq), 124.3, 126.5 (Cq), 128.1, 129.4, 132.4, 132.9, 135.8 (Cq), 136.2, 137.6 (Cq), 183.1 (C=O) ppm. MS: (70 eV) m/z (%) = 368/366 (2/2) [M]+ •, 287 (9), 158 (100), 130 (17), 83 (61). Anal. Calcd. For C19H15BrN2O (367.25): C, 62.14; H, 4.12; N, 7.63. Found. C, 62.33; H, 4.11; N, 7.99.
2-(2-Bromobenzyl)-3-(1H-indol-3-yl)-3-oxopropanenitrile (5e). This compound was obtained according to the above procedure from 4e (0.500 g) as a white solid, mp 246 °C. FTIR (ATR): v = 3259 (N-H), 3028, 2247 (CN), 1666 (C=O), 1579 cm1. 1H NMR (400 MHz, DMSO-d6): δ = 3.34-3.37 (m, 2H), 5.06 (t, 1H), 7.21-7.29 (m, 3H), 7.38 (t, 1H), 7.52 (t, 2H), 7.64 (d, 1H), 8.18 (d, 1H), 8.61 (s, 1H), 12.27 (s, 1H) ppm. 13 C NMR (100 MHz, DMSO-d6): δ =35.4 (CH2), 39.9 (CH), 112.5, 113.7 (Cq), 118.4 (CN), 121.5, 122.5, 123.6, 124.2 (Cq), 125.4 (Cq), 127.8, 128.4, 129.3 (Cq), 131.6, 132.7, 136.0 (Cq), 136.8, 184.3 (C=O) ppm. MS: (70 eV) m/z (%) = 354/352 (2.9/2.5) [M]+ •, 144 (100), 273 (43), 116 (23), 89 (13).
2-(1-Benzyl-1H-indol-3-yl)-2-(2-bromobenzyl)-3-oxopropanenitrile (5f). This compound was obtained according to the above procedure from 4f (0.300 g) as a white solid, mp 146 °C. FTIR (ATR): v = 3059 (C-H), 2250 (CN), 1671 (C=O), 1568 cm1. 1H NMR (400 MHz, CDCl3): δ = 3.34 (dd, J = 13.7, 9.8 Hz, 1H), 3.55 (dd, J = 13.6, 6.0 Hz, 1H), 4.50 (dd, J = 9.7, 6.0 Hz, 1H), 5.29 - 5.45 (m, 2H), 7.12 - 7.27 (m, 3H), 7.33 - 7.47 (m, 8H), 7.56 (dd, J = 8.0, 1.2 Hz, 1H), 7.89 (s, 1H), 8.42 - 8.50 (m, 1H) ppm. 13C NMR (100 MHz, CDCl3): 5 = 37.1 (CH2), 40.7 (CH), 51.1 (CH2Ph), 110.4, 114.5 (Cq), 117.6 (CN), 122.8, 123.7, 124.2 (Cq), 124.4, 126.7 (Cq), 127.6, 128.1, 128.7, 129.3, 129.4, 132.4, 132.9, 134.7 (Cq), 135.3, 135.6 (Cq), 137.3 (Cq), 183.6 (C=O) ppm. MS: (70 eV) m/z (%)= 444/442 (8/8) [M]+ •, 234 (15), 149 (8), 91 (100). Anal. Calcd. For C25H19BrN2O (443.34): C, 66.44; H, 4.24; N 6.87. Found. C, 66.61; H, 4.33; N, 6.91.
3-(1-Benzyl-1H-indol-3-yl)-2-((2-chloroquinolin-3-yl)methyl)-3-oxopropanenitrile (5g). This compound was obtained according to the above procedure from 4g as a white solid, mp 198 °C. 1H NMR (400 MHz, CDCl3): δ = 3.44 (dd, J = 13.8, 9.3 Hz, 1H), 3.70 (dd, J = 13.5, 6.1 Hz, 1H), 4.63 - 4.53 (m, 1H), 5.34 (s, 2H), 7.24 - 7.15 (m, 2H), 7.43 -7.28 (m, 6H), 7.57 (t, J = 7.4 Hz, 1H), 7.73 (t, J = 7.5 Hz, 1H), 7.82 (d, J = 8.1 Hz, 1H), 7.89 (s, 1H), 8.00 (d, J = 8.4 Hz, 1H), 8.23 (s, 1H), 8.41 (dd, J = 8.3, 1.5 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 34.3 (CH2), 40.3 (CH), 51.2 (CH2Ph), 110.5, 114.3 (Cq), 117.5 (Cq), 122.7, 123.8, 124.5, 126.6 (Cq), 127.3 (Cq), 127.5, 127.6, 127.7, 127.9 (Cq), 128.1, 128.8, 129.3, 130.8, 134.6 (Cq), 135.4, 137.3 (Cq), 140.6, 147.1(Cq), 150.1 (Cq), 182.8 (C=O) ppm. MS: (70 eV) m/z (%) = 451/449 (0.1/0.3), 234 (14), 91 (100).
2-((2-Chloroquinolin-3-yl)methyl)-3-(1-methyl-1H-indol-3-yl)-3-oxopropanenitrile (5h). This compound was obtained according to the above procedure from 4h as a white solid, mp 210 °C. FTIR (ATR): v = 2987, 2943, 2910 (C-H), 2243 (CN), 1663 (C=O), 1537 (C=C) cm-1. 1H NMR (400 MHz, CDCl3): δ = 3.48 (dd, J = 14.0, 9.2 Hz, 1H), 3.75 (dd, J = 13.0, 4.6 Hz, 1H), 3.86 (s, 3H), 4.62 (dd, J = 9.1, 6.3 Hz, 1H), 7.36 (d, J = 3.7 Hz, 3H), 7.56 (t, J = 7.5 Hz, 1H), 7.73 (t, J = 7.2 Hz, 1H), 7.82 (d, J = 8.2 Hz, 1H), 7.88 (s, 1H), 8.00 (d, J = 8.5 Hz, 1H), 8.23 (s, 1H), 8.38 (dd, J = 6.6, 2.8 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 33.8 (NCH3), 33.9 (CH2), 39.8 (CH), 110.0, 114.0 (Cq), 117.7 (Cq), 122.6, 123.7, 124.4, 126.4 (Cq), 127.3(Cq), 127.5, 127.6, 128.1(Cq), 128.2, 130.8, 136.2, 137.6 (Cq), 140.6, 147.2(Cq), 150.2 (Cq), 182.3 (C=O) ppm. MS: (70 eV) m/z (%) = 375/373 (0.1/0.3), 158 (100), 77 (29).
3-(1-Butyl-1H-indol-3-yl)-2-((2-chloroquinolin-3-yl)methyl)-3-oxopropanenitrile (5i). This compound was obtained according to the above procedure from 4i as a white solid, mp 154 °C. FTIR (ATR): v = 3022, 2959, 2883 (C-H), 2258 (CN), 1678 (C=O), 1567 (C=C) cm-1. 1H NMR (400 MHz, CDCl3): δ = 0.96 (t, J = 6.8 Hz, 3H), 1.41 - 1.28 (m, 2H), 1.92 - 1.77 (m, 2H), 3.49 (dd, J = 13.1, 9.0 Hz, 1H), 3.75 (dd, J = 13.8, 5.4 Hz, 1H), 4.16 (t, J = 6.5 Hz, 2H), 4.69 - 4.50 (m, 1H), 7.43 - 7.30 (m, 3H), 7.57 (t, J = 6.9 Hz, 1H), 7.73 (t, J = 7.4 Hz, 1H), 7.82 (d, J = 7.9 Hz, 1H), 7.91 (s, 1H), 8.02 (d, J = 8.2 Hz, 1H), 8.25 (s, 1H), 8.43 - 8.32 (m, 1H) ppm. 13C NMR (100 MHz, CDCl3): δ = 13.6 (CH3), 20.1 (CH2), 31.7 (CH2), 33.9 (CH2), 39.9 (CH), 47.3 (CH2), 110.3, 113.9 (Cq), 117.7 (Cq), 122.7, 123.6, 124.3, 126.6, 127.6, 127.7, 128.1, 128.1 (Cq), 130.9, 135.3 (Cq), 136.4 (Cq), 137.0 (Cq), 140.7, 147.1 (Cq), 150.1 (Cq), 182.5 (C=O) ppm; MS: (70 eV) m/z (%)= 415 (5), 380(21), 200 (100), 144(26).
Methyl 2-cyano-3-(4-methoxyphenyl)propanoate (13). This compound was obtained according to the above procedure from 12 (0.150 g) as a colorless oil. FTIR (ATR): v = 3008 (C-H), 2232 (CN), 1721 (C=O) cm-1. 1H NMR (400 MHz, CDCl3): δ = 3.36 - 3.03 (m, 2H), 3.73 (dd, J = 8.3, 5.7 Hz, 1H), 3.83 (d, J = 3.9 Hz, 6H), 6.90 (d, J = 8.3 Hz, 2H), 7.22 (d, J = 8.2 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 35.1 (CH2), 39.8 (CH), 53.9 (OCH3), 55.3 (OCH3), 114.3, 116.1 (CN), 127.2 (Cq), 130.1, 159.2 (Cq), 166.1 (C=O) ppm.
2-Cyano-N-(2-iodophenyl)-3-phenylpropanamide (15). This compound was obtained according to the above procedure from 14 (0.120 g) as a white solid, mp 224 °C. FTIR (ATR): v = 3223 (N-H), 3068, 2221 (CN), 1662 (C=O), 1568 cm1. 1H NMR (400 MHz, DMSO-d6): δ = 3. 22 (dd, J = 13.6, 9.0 Hz, 1H), 3.40 (dd, J = 13.3. 8.8 Hz, 1H), 4.36 (dd, J = 9.0, 6.4 Hz, 1H), 7.06 (td, J = 7.6, 1.7 Hz, 1H), 7.48 - 7.26 (m, 7H), 7.92 (dd, J = 7.8, 1.4 Hz, 1H), 10.11 (s, 1H) ppm. 13C NMR (100 MHz, DMSO): δ = 36.1 (CH2), 40.4 (CH), 97.1 (Cq), 118.3 (CN), 127.7, 128.0, 128.9, 129.0, 129.3, 129.6, 137.0 (Cq), 139.0 (Cq), 139.6, 164.3 (C=O) ppm.
The synthesis of the remaining known compounds have previously been reported elsewhere.
Results and discussion
Searching for diverse precursors to be used as starting material for the synthesis of new heterocyclic compounds, we required to obtain a variety of α,β-reduced ketone- and ester-derivatives. Selective reduction of the C=C double bond from their corresponding α,β-unsaturated precursors was chosen as the more expeditious approach. Firstly, looking for a simple, cheap, selective and quantitative gram-scale yield of the desired partially-reduced ketone 2a,Scheme 1, we performed an adaptation of the Cornuberts procedure for the selective reduction of chalcone 1a as model reaction, using chloroform as catalyst-poisoning agent (Cornubert & Eggert, 1954; Cornubert & Thomas, 1954). Despite several synthetic variants were evaluated (i.e. adjusting the Raney-Nickel catalyst ratio, reaction times, volume of chloroform used as poisoning agent and hydrogen pressure, among others), not much success was achieved. Mixtures of the expected product 2a along with the undesired doubly reduced product 3a were obtained in all cases.
Finally, we found that controlling the volume of EtOH used as solvent was critical to achieve better results. Thus, in our alternative approach the minimum as possible volume of EtOH was used to dissolve the starting chalcones 1a-d. Afterwards, the catalytic hydrogenation of the saturated ethanolic solutions of 1a-d in the presence of Raney-Nickel poisoned with CHCl3 at room temperature under (20 psi) pressure, afforded the desired ketones 2a-d in almost quantitative yields, table 1. A gentle warming was required in some cases to get saturated solutions of chalcones 1. Alternatively, the use of DCM instead CHCl3 also worked well.
Table 1 Selective reduction of the α,β-unsaturated C=C double bonds in chalcones 1. a,b
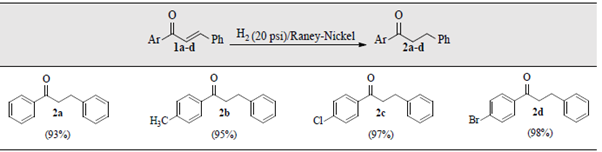
a Reaction conditions: Chalcone 1 (1.0 gram), Raney-Nickel (0.3 g), EtOH (5-10 mL), CHCl3 or DCM (0.5 mL), rt, 15-20 min. bFor more details see Materials and Methods section.
As mentioned above, we discovered that the control of the volume of solvent (i.e. EtOH) dramatically improved the selectivity towards the partially hydrogenated keto-derivatives 2, in comparison with the previous approaches where larger volumes of EtOH were used (Cornubert & Eggert, 1954; Cornubert & Thomas, 1954; Tucker, 1950). This finding is supported by the experimental fact that products 2 were less soluble in the reaction media than the starting chalcones 1. Thus, products 2 precipitated from the saturated ethanolic solutions as quickly as they were formed avoiding the subsequent reduction of their carbonyl groups. To confirm the above suggestion, a controlled experiment using chalcone 1a and a large volume of EtOH was performed. In this case no solid precipitated and contrary formation of the dense-oily saturated alcohol 3a turned into the main product of the reaction mixture.
At this stage, it is worth to mention that attempts to extend the above approach to Knoevenagel cyano-keto derivatives 4 did not work. Alternatively, after several assays we found that trying the Knoevenagel adducts 4 with NaBH4 in a _p-dioxane/MeOH mixture at 70 oC afforded the expected partially-reduced products 5 in satisfactory yields, table 2.
Table 2 Selective reduction of the C=C double bonds in α,β-unsaturated α-cyanoketones 4. a,b,c
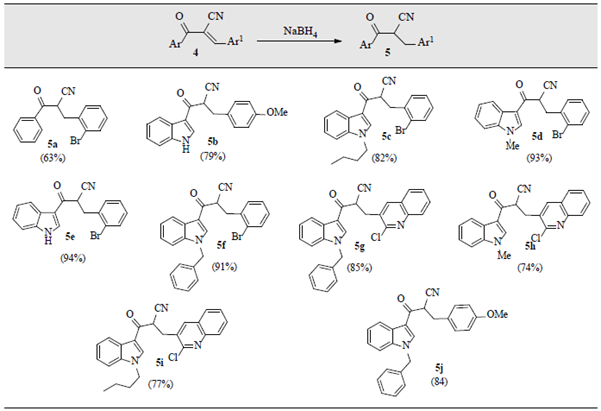
a Reaction conditions: Knoevenagel adduct 4 (0.2-1.0 gram, 1 equiv.), NaBH4 (1 equiv.), p-dioxane/MeOH (1:4) (5-10 mL), 70 oC. bFor more details see Materials and Methods section. c The synthesis of the starting indoles and cyanoindoles as precursors for cyanochalcones 4b-j is described in the Materials and Methods section.
Structures of the new obtained compounds 4 and 5 were confirmed by 1H NMR, 13C NMR, and MS spectra. Particularly, a comparative analysis of the 13C NMR spectra for the starting α-cyanoketone 4d and its corresponding partially-reduced product 5d confirmed the efficacy of the established NaBH4-metiated selective-reductive conditions, figure 1. The appearing of two new aliphatic carbon atom signals at high field (i.e. 40.36 and 36.5 ppm) in spectrum for 5d, the disappearing of the =C signal at 152.21 ppm in the spectrum for 4d and the presence of the carbonyl (C=O) signals in both spectra at (178.6 and 183.1) ppm for 4d and 5d, respectively, are in agreement with the selectivity of the reduction process.
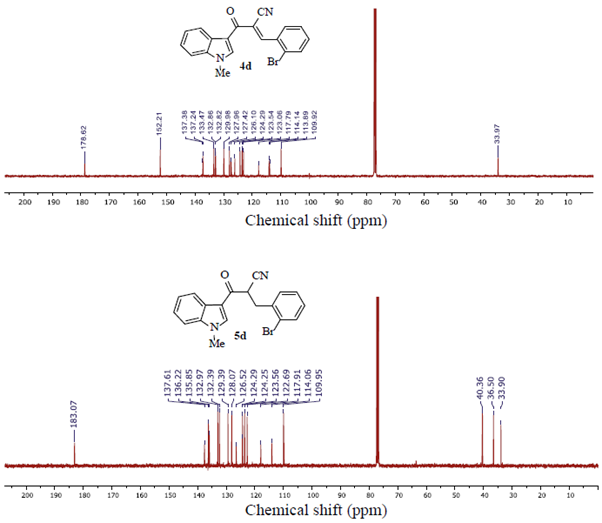
Interestingly, when the above reactions were performed at room temperature mixtures of 5 and their corresponding doubly reduced side-hydroxyl products 11 (i.e. alcohol analogues type 3), were obtained. A suggested mechanistic sequence for the selective formation of products 5 and alternatively, for the double reduced side-hydroxyl products 11 is drawn in Scheme 2.
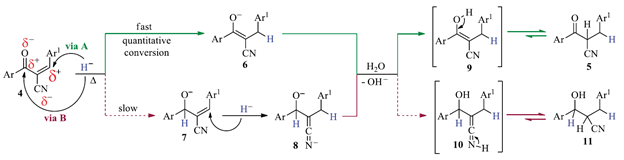
Scheme 2 Suggested mechanistic sequence for the selective formation of the partially-reduced products 5
In principle, the presence of two electron-withdrawing functional groups (i.e. CN and C=O) is the responsible of the higher electrophilicity of the β-carbon atom than the carbonyl C=O group. Apparently, a higher temperature also increases the electrophilicity of such β-carbon atom in 4, as well as, the reactivity of the hydride anion (i.e. less selectivity), for a faster Michael type addition of H- (via A) than that for the carbonyl H- addition (via B), affording preferably the enolate species 6. This species should prevent the subsequent reduction of the carbonyl group toward the hydroxyl-compound 11 as happened in the formation of alcoholic derivatives 3, showed in Scheme 1. Then, the work-up by adding water to the reaction crude (after removing the solvent under reduced pressure), supplied the required protons to recover selectively the target ketonic tautomer 5. We assume that using lower temperatures reduces the electrophilicity of the β-carbon atom in 4 and hence, increases the chance that H- attack the p-carbon atom but also the carbonyl group in 4 through a mixture of species 6 and 7, respectively. A further p-addition of H- over species 7 (through species 8), followed by the usual aqueous work-up afforded the detected and isolated mixture of products 5 and the doubly reduced cianohydroxyl side-product 11.
Remarkably, the established approach showed to be robust and a miscellaneous of cyanoester 12, cyanamide 14, ketoester 16, diester 18 and diketone 20 derivatives, were also selectively reduced, as shown in tables 3and4.
Table 3 Selective reduction of the C=C double bonds in α,β-unsaturated α-cyanoester 12 andα-cyanamide 14. a,b
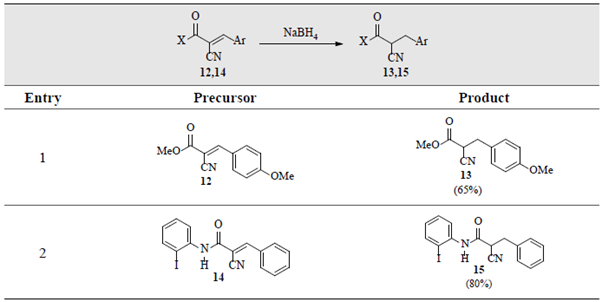
a Reaction conditions: α-cyano-derivatives 12/14 (0.2-1.0 gram, equiv.), NaBH4 (1 equiv.), p-dioxane/MeOH (1:4) (5-10 mL), 70 oC. bFor more details see Materials and Methods.
Table 4 Selective reduction of the C=C double bonds in α,β-unsaturated α-ketoester 16, 1,3-diester 18 and 1,3-diketone 20. a,b
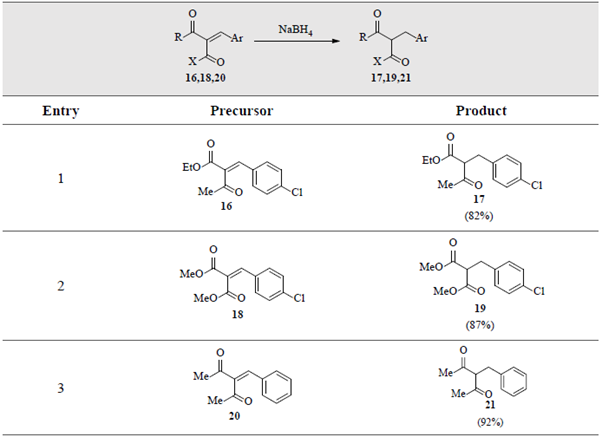
a Reaction conditions: Dicarbonyl compounds 16/18/20 (0.2-1.0 gram, 1 equiv.), NaBH4 (1 equiv.), p-dioxane/MeOH (1:4) (5-10 mL), 70 oC. b For more details see Materials and Methods.
Noteworthy, when this approach was applied to simple Claisen-Schmidt condensation products (i.e. chalcones 1e, 23, 25 and bis-chalcone 27), their corresponding hydroxyl-derivatives 22, 24, 26 and 28, respectively, were obtained as unique isolated products, without affecting their C=C double bonds, table 5.
Table 5 Selective reduction of the C=O double bond in α,β-unsaturated ketones 1e, 23, 25 and 27. a,b
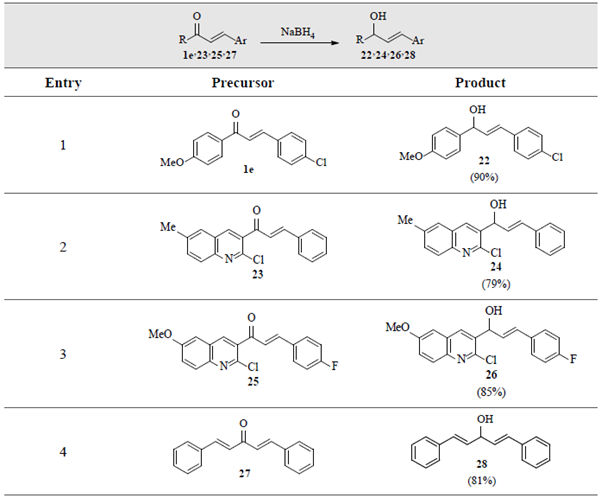
a Reaction conditions: Chalcone 1e, 23, 25 or 27 (0.2-1.0 gram, 1 equiv.), NaBH4 (1 equiv.), p-dioxane/MeOH (1:4) (5-10 mL), 70 oC. b For more details see Materials and Methods.
A suggested mechanistic sequence for the selective reduction of the carbonyl (C=O) groups for the above simple ketonic devivatives A (i.e. 1e, 23, 25 and 27) is drawn in Scheme 3.
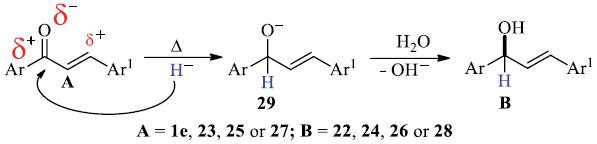
Scheme 3 Suggested mechanistic sequence for the selective reduction of the carbonyl (C=O) groups in simple ketonic devivatives A (i.e 1e, 23, 25 and 27)
In this case, the absence of the second electron-withdrawing group in simple ketones A, in comparison with precursors 4, causes a lower electrophilicity of the p-carbon atom than that of the carbonyl C=O carbon atom in structure A. In this sense, the nucleophilic attack by H- ion is produced preferably over the carbonyl C=O carbon atom producing the alkoxy intermediate 29. Subsequently, by performing the work-up as usual, afforded the corresponding isolated allylic alcohols B (i.e. 22, 24, 26 or 28) as shown in Scheme 3.
In summary, we implemented two complementary approaches for the selective reduction of the C=C functionality in diverse α,β-unsaturated precursors. In general, poisoned Raney-Nickel-mediated catalytic hydrogenation under controlled volume of EtOH worked well for ordinary chalcones 1 , while warm p-dioxane/MeOH/NaBH4-mediated chemical reduction was the best reductive conditions for Knoevenagel condensation adducts 4, 12, 14, 16, 18 and 20, affording the respective partially reduced products in almost quantitative to excellent yields. The fact that it was possible to reduce, in a selective way, the carbonyl group on some chalcone derivatives is also highlighted in this approach. Moreover, trying to explain the selective NaBH4-mediated reductive process a plausible mechanistic pathway was proposed in this regard.

