











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkPortada de Science
El Consorcio Telómero a Telómero (T2T) ha completado la secuencia de un desafiante 8% del genoma humano que quedó sin resolver en el Proyecto Genoma Humano. En esta visualización, cada cromosoma comienza en la parte inferior derecha y se envuelve alrededor de un centro tácito, con los cromosomas X y 1 a 22 dispuestos de afuera hacia adentro (el cromosoma Y no se se muestra). Las regiones recién completadas se resaltan en rojo.
Gráfico: V. Altounian/Ciencia Datos: Consorcio T2T
Sergey Nurk, Sergey Koren, Arang Rhie, Mikko Rautiainen, et al. (2022). The complete sequence of a human genome. Science 2022, Vol. 376, No. 6588, pp. 44-53. https://doi.org/10.1126/science.abj6987
Hace más de 20 años, en febrero 15 y 16 del año 2001, las revistas Nature y Science publicaron la secuencia de nucleótidos que muchos llamaron "completa" del genoma humano (International Human Genome Sequencing Consortium, 2001; Venter et al., 2001). Sin embargo, lo que se reportó fue apenas una primera aproximación a la totalidad del genoma que cubría cerca del 92% de las secuencias de individuos seleccionados, correspondiente a la fracción cromosómica que se asocia a la eucromatina que se tiñe más fácilmente con coloraciones para ADN por estar más compactada. Se dejó por fuera la fracción restante del 8% (heterocromatina), compuesta por secuencias típicamente repetitivas consideradas en ese entonces genéticamente inactivas (llamadas satelitales), localizadas tanto en los extremos de los cromosomas, o telómeros, como alrededor de los centrómeros -la parte del cromosoma por donde el huso mitótico o meiótico tracciona los cromosomas para separarlos en las dos nuevas células hijas-.
Hace unos días, el 1 de abril de 2022, cerca de 22 años después de haberse concluido en los laboratorios los análisis que permitieron esta primera aproximación, el consorcio T2T (sigla que remite en inglés a Telomere-to-Telomere), publicó en la revista Science lo que se tituló propiamente como la "Secuencia completa de un genoma humano" (Nurk et al., 2022). Y este título se refiere a "un" solo genoma humano, porque esta secuencia completa se hizo a partir del ADN de una célula particular de una mujer que la desarrolló en su placenta sin el aporte de uno de los genomas parentales. Este tipo de célula puede convertirse en una Mola Hidatiforme Completa (o CMH, por su acróstico en inglés) y no es viable. Generalmente la CMH es resultado de la pérdida del componente materno en el ADN nuclear con la duplicación del complemento paterno, resultando, en este caso, en un cariotipo homocigoto 46, XX (como si fuera de una mujer), producto de una duplicación simple de la información contenida en 23 cromosomas de un solo individuo: gracias a esta estrategia se logró la clave técnica que permitió analizar la secuencia de este genoma con mayor facilidad. Sin embargo, debe tenerse en cuenta que el genoma reportado como T2T-CMH13 por este equipo de 100 investigadores provenientes de Estados Unidos, Rusia, Alemania, Croacia y Gran Bretaña incluye información sobre 23 cromosomas correspondientes a la serie completa de los autosomas (del 1 al 22) y a un solo cromosoma sexual (el cromosoma-X) en calidad de cromosoma 23: así es que este "genoma completo" no incluye aún información sobre el cromosoma-Y asociado al género masculino.
En todo caso, este reporte trae información sobre más de 3.400 genes previamente desconocidos (79 de los cuales parecen ser codificantes) y, como lo muestra un extracto de la tabla 1 original de esta publicación, porcentajes variables de diferentes marcadores genéticos repetitivos o satelitales.
Tabla 1 (extracto). Comparación de las secuencias GRCh38 (4)1 y T2T-CMH13
| Descriptor | GRCh38 | T2T-CMH13 | Diferencia (%) |
|---|---|---|---|
| Número de genes | 60.090 | 63.494 | + 5,7 |
| Codificantes | 19.890 | 19.969 | + 0,4 |
| Repeticiones (%) | 51,89 | 53,94 | [+ 2,05] |
| Bases repetidas (Mbp) | 1.516,37 | 1.647,81 | + 8,7 |
| Elementos nucleares largos espaciados (LINE) | 626,33 | 631,64 | + 0,8 |
| Elementos nucleares cortos espaciados (SINE) | 386,48 | 390,27 | + 1,0 |
| Repeticiones largas terminales (LTR) | 267,52 | 269,91 | + 0,9 |
| Satélites | 76,51 | 150,42 | + 96,6 |
| ADN (DNA) | 108,53 | 109,35 | + 0,8 |
| Repeticiones simples | 36,57 | 77,69 | + 112,9 |
| Repeticiones de baja complejidad | 6,16 | 6,44 | + 4,6 |
| Retroposones | 4,51 | 4,65 | + 3,3 |
| ARN ribosomal (rRNA) | 0,21 | 1,71 | [+ 715] |
1 Secuencia genómica humana actualizada producida por el Genome Reference Consortium con base en las secuencias publicadas en 2001 y sucesivamente actualizadas (Schneider et al., 2020).
Además de esta proeza científica, cuyos aspectos principales se incluyen en el extracto de la figura 1 original, la misma edición de la revista Science de abril 1 de este año trae una sección especial con diferentes consideraciones y reportes complementarios como el de Gershman et al. (2022), con el análisis de los patrones "epigenéticos" de esta secuencia genómica: es decir, lo que va más allá de la serie de nucleótidos reportada por el T2T en un estudio supragénico de alta resolución con las modificaciones no nucleotídicas que se presentan en las regiones correspondientes al 8% revelado en los brazos cortos de cromosomas acrocéntricos enteros, telómeros y diversas clases de repeticiones. Este análisis proporciona un marco que permitirá investigar la regulación de las regiones más difíciles del genoma humano, proporcionando información sobre los determinantes epigenéticos que controlan la actividad genética y la función celular.
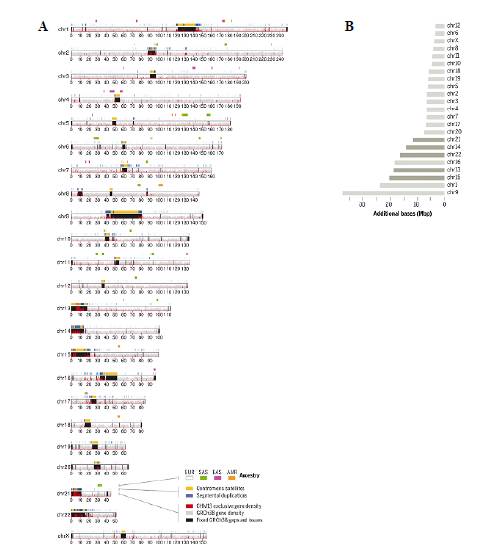
Figura 1 (extracto). Ideogramas de los 23 cromosomas secuenciados en el reporte del T2T. (A) Características del ensamblaje para cada cromosoma (chr). (B) Nucleótidos adicionales en el ensamblaje CHM13 en relación con el ensamblaje GRCh38 en cada cromosoma, incluyendo los cromosomas acrocéntricos resaltados en gris oscuro.
Para terminar, debe considerarse muy especialmente que el genoma reportado corresponde a un solo individuo de origen europeo con trazas de genomas neandertales y muy pocas regiones compartidas con comunidades actuales de los demás continentes del planeta. Así, es de esperar que este genoma de referencia (T2T-CMH13) sirva, como dicen los autores al final de su artículo, para "revelar la diversidad completa del género humano" a través de iniciativas como la del Human Pangenome Reference Consortium (Miga & Eang, 2021).

