










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombia Médica
On-line version ISSN 1657-9534
Colomb. Med. vol.38 no.3 Cali July/Sept. 2007
Complejo agnatia holoprosencefalia: informe de caso
Fernando Suárez-Obando M.D.*, Juan Carlos Prieto, M.D., M.Sc.*
1. Instituto de Genética Humana, Pontificia Universidad Javeriana, Bogotá, D.C., Colombia.
e-mail: fernando.suarez@javeriana.edu.co jcprieto@javeriana.edu.co
Recibido para publicación febrero 14, 2007 Aceptado para publicación julio 4, 2007
RESUMEN
Se presenta un caso de complejo agnatia holoprosencefalia y se realiza una revisión de la literatura, en relación con la compleja etiología genética y embriológica de este conjunto de malformaciones mayores de la cara y el sistema nervioso central. Se trata del primer caso que se informa en la literatura colombiana.
Palabras clave: Genética; Holoprosencefalia; Anomalías craneofaciales.
Agnathia holoprosencephaly complex: case report
SUMMARY
A case report of the agnathia holoprosencephaly complex and a review of the literature related to the complex genetic and embryologic aetiology of this group of major birth defects of face and central nervous system are informed. The present clinical case is the first reported in Colombia.
Keywords: Genetics; Holoprosencephaly; Craniofacial anomalies.
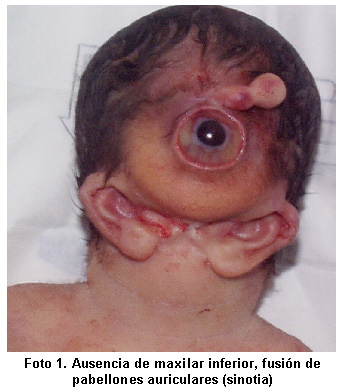
El complejo agnatia holoprosencefalia, es una malformación congénita mayor, casi siempre letal, caracterizada por ausencia o hipoplasia severa del maxilar inferior, posición anormal de los pabellones auriculares y holoprosencefalia1. Se presenta un caso de agnatia holoprosencefalia, asociado con ciclopía, sinoftalmia, probóscide y sinotia. La baja prevalencia de esta entidad y la extrema complejidad de la malformación dan relevancia a este informe clínico, el primero en Colombia.
El estudio de esta anomalía sobresale en el contexto clínico y embriológico, debido a que su estudio explica las consecuencias de la falta de segmentación y desarrollo embriológico del sistema nervioso central, además de ser un cuadro clínico rico en semiología médica.
INFORME DE CASO
Recién nacido muerto de 36 semanas de edad gestacional, de madre de 28 años de edad (padres no consanguíneos), producto de segundo embarazo sin exposición a teratógenos. Control prenatal normal, hasta la semana 30, cuando a través de ecografía obstétrica se identifica, ventriculomegalia, agenesia del cuerpo calloso, posición anormal de los pabellones auriculares y retrognatia. La madre consulta por ausencia de movimientos fetales en la semana 36 de gestación, se realiza una nueva ecografía que ratifica los hallazgos anteriores y se diagnostica óbito fetal; parto por cesárea iterativa.
Al examen físico se encuentra: Peso, 2300 g; talla, 48 cm. Perímetro cefálico, 39 cm. Presencia de probóscide central, ciclopía, y ausencia de apertura bucal (astomia), agnatia (ausencia de maxilar inferior) y fusión de pabellones auriculares en la línea media o sinotia (signo clínico también denominado otocefalia) (Foto 1). El resto del examen físico fue normal. Cariotipo de 550 bandas, bandeo G, 46, XY, sin alteraciones estructurales ni numéricas. Previa firma del consentimiento informado, según se registra en la historia clínica, se llevó a cabo el estudio fotográfico y post-mortem; en éste el sistema nervioso central reveló agenesia de cuerpo calloso, sin fusión talámica, ventrículo único e hipoplasia de cerebelo confirmado una holoprosecenfalia alobar, en el macizo facial se evidenció esbozo de lengua, globo ocular único, y fusión total de nervio óptico.
DISCUSIÓN
El complejo agnatia holoprosencefalia, o complejo disgnatia (OMIM: 202650)2, se caracteriza por hipoplasia severa o agenesia del maxilar inferior (agnatia) y presencia de pabellones auriculares ubicados anterior e inferiormente. La gama de anormalidades asociadas incluye sinotia (fusión de los pabellones auriculares en la línea media) y la holoprosencefalia. El conjunto de la agnatia y sinotia se denomina otocefalia3. Una apertura bucal muy pequeña o ausente es una característica constante, incluso puede haber persistencia de la membrana bucofaríngea. Debido a la obstrucción respiratoria y a las malformaciones del sistema nervioso central, casi todos los afectados fallecen en el período perinatal.
ETIOLOGÍA Y DESARROLLO EMBRIONARIO
Pauli et al.4, informan este complejo en dos hermanas, que fallecieron en el período perinatal, y sugieren una herencia autosómica recesiva; sin embargo, el análisis cromosómico prometafásico posterior de las dos niñas y de sus padres, estableció que el padre era portador de una translocación equilibrada y que una de las fallecidas tenía una translocación desequilibrada, siendo uno de los puntos de ruptura, el locus para la holoprosencefalia ubicado en 18p. Krassikoff y Sekhon5, comunicaron otro caso familiar donde tres afectados, eran portadores de una duplicación de 6p y monosomía de 18p. El padre de los afectados así como su madre y hermano portaban una translocación equilibrada t (6:18). Los hallazgos de anormalidades cromosómicas, sugerían que la herencia no era recesiva y que el o los genes comprometidos tenían sus loci cerca de los relacionados con el locus de la holoprosencefalia, según lo insinuaba la presentación clínica.
Erlich et al.6 observaron la transmisión de madre a hija del complejo disgnatia, pues la madre hizo una presentación clínica leve, que apuntaba a una transmisión dominante con expresividad variable. También proponían que las bases moleculares de este trastorno eran las alteraciones en el gen OTX2 (sigla en inglés para orthodenticle drosophila homolog of 2), gen homeótico ubicado en 14q21-q22, que se expresa en las regiones dorsales y ventrales del telencéfalo, diencéfalo y mesencéfalo7.
Guion-Almeida et al.8 propusieron que los pacientes descritos por Erlich et al.6, en realidad sufrían el síndrome aurículo-condilar (OMIM: 602483)9, que consiste en severa micrognatia y anormalidades auriculares, sin anomalías en el sistema nervioso central. Esta sugerencia no se ha podido confirmar, debido a que ambos síndromes pueden hacer parte de un mismo cuadro de la enfermedad, como lo mencionan otras descripciones clínicas en las que se ha clasificado al complejo en dos tipos: una forma severa que incluye la holoprosencefalia y otra leve en la que no hay anormalidades del sistema nervioso central (10,11).
El modelo murino del complejo agnatia holoprosencefalia, demuestra que el fenotipo surge de mutaciones en el gen OTX2, en estado heterocigoto y que la severidad depende de otros genes modificadores en distintos loci (12), sugiriendo nuevamente un mecanismo de herencia dominante de expresividad variable.
Se ha postulado que los efectos de las mutaciones del gen OTX2 y de sus genes modificadores, son la disminución de la capacidad inductiva del mesodermo precordial, que lleva a la agnatia por una migración neuronal anómala hacia las porciones ventrales del primer arco branquial y la segunda bolsa faríngea, fenómenos que suceden entre los 22 y 26 días de edad gestacional (13). La expresión del gen OTX2, quizá tenga relación con la vía de señalización Sonic Hedgehog, que es una familia de proteínas de señalización intercelular que juegan un papel primordial en el desarrollo embrionario; las mutaciones en esta vía generan diversos fenotipos que incluyen la holoprosencefalia lobar, alobar y semilobar, malformaciones que pueden cursar con cebocefalia, etmocefalia o probóscide (14).
En el caso que aquí se informa, es obvio que la induc-ción mesodérmica se vio severamente afectada en los arcos branquiales, y que por esta razón la implantación auricular fue en la línea media (sinotia), debido a la ausencia del maxilar inferior. Otro aspecto llamativo del caso, es la ciclopía, que tiene la particularidad de ser un solo ojo en una sola orbita bien definida, lo que la diferencia de la ciclopía secundaria a la sinoftalmía o fusión ocular, donde las estructuras oculares y la bóveda orbital no están tan bien definidas. La ciclopía o sinoftalmía, son secundarias a la pérdida de estructuras mediales de la holoprosencefalia.
De tal modo que el defecto inicial pudo generarse en anomalías del desarrollo de las células que se derivan de las crestas neurales cefálicas por mutaciones en el gen OTX2, seguido de una inducción mesodérmica anormal en los arcos branquiales, ausencia del desarrollo en las estructuras de la línea media del cerebro secundario a falla en la estimulación del tubo neural y la consecuente ciclopía y probóscide, componentes del conjunto de anormalidades faciales de la holoprosencefalia.
FRECUENCIA Y ASESORÍA GENÉTICA
Tanto la complejidad como la severidad de las malformaciones pueden explicar su baja frecuencia, debido a que la mayoría de defectos de este tipo generan la interrupción temprana del embarazo. Hubo dos casos entre 200,000 nacimientos en el estudio colaborativo español de malformaciones congénitas (ECEMC); es decir, se indica una prevalencia de 1 en 100,000 casos (14), y alrededor de 80 casos se han comunicado en la literatura (15). Este es el primer ejemplo del complejo agnatia holoprosencefalia que se informa en Colombia.
Aunque no hay consenso absoluto sobre su patrón de herencia, el riesgo de recurrencia en una pareja completamente sana sin anomalías cromosómicas es menor de 1%; en este caso los padres no eran portadores de anormalidades cromosómicas ni afectados por holoprosencefalia lobar o semilobar.
CONCLUSIONES
El complejo agnatia holoprosencefalia constituye un grupo de malformaciones severas que compromete el desarrollo del sistema nervioso central y de los arcos branquiales; casi siempre es incompatible con la vida y su extrema complejidad puede explicar su baja frecuencia.
REFERENCIAS
1. Carey J. External ear. En: Stevenson R, Hall J, Goodman R (eds.). Human malformations and related anomalies. New York: Oxford University Press; 1993. p. 193-219. [ Links ]
2. Online Mendelian Inheritance in Man, OMIM (TM). Baltimore: Johns Hopkins University. MIM Number: 202650. (fecha de acceso: septiembre 20, 2006. URL disponible: http://www.ncbi.nlm.nih.gov/sites/entrez?db=OMIM [ Links ]
3. O'neill BM, Alessi AS, Petti NA. Otocephaly or agnathia-synotia-microstomia syndrome: report of a case. J Oral Maxillofac Surg 2003; 61: 834-837. [ Links ]
4. Pauli RM, Pettersen JC, Arya S, Gilbert EF. Familial agnathia-holoprosencephaly. Am J Med Genet 1983; 14: 677-698. [ Links ]
5. Krassikoff N, Sekhon GS. Familial agnathia-holoprosencephaly caused by an inherited unbalanced translocation and not autosomal recessive inheritance. Am J Med Genet 1989; 34: 255-257. [ Links ]
6. Erlich MS, Cunningham ML, Hudgins L. Transmission of the dysgnathia complex from mother to daughter. Am J Med Genet 2000; 95: 269-274. [ Links ]
7. Akagi T, Mandai M, Ooto S, Hirami Y, Osakada F, Kageyama R, et al. Otx2 homeobox gene induces photoreceptor-specific phenotypes in cells derived from adult iris and ciliary tissue. Invest Ophthalmol Vis Sci 2004; 45: 4570-4575. [ Links ]
8. Guion-Almeida ML, Zechi-Ceide RM, Vendramini S, Kokitsu-Nakata NM. Auriculo-condylar syndrome: additional patients. Am J Med Genet 2002; 112: 209-214. [ Links ]
9. Online Mendelian Inheritance in Man (OMIM) (TM). Baltimore: Johns Hopkins University. MIM Number: 602483. (fecha de acceso abril 12, 2006). URL disponible en: http://www.ncbi.nlm.nih.gov/omim/ [ Links ]
10. Bixler D, Ward R, Gale DD. Agnathia-holoprosencephaly: a developmental field complex involving face and brain. Report of 3 cases. J Craniofac Genet Dev Biol 1985; 1 (Suppl): 241-249. [ Links ]
11. Kamiji T, Takagi T, Akizuki T, Kurukata M, Ohmori K. A long surviving case of holoprosencephaly agnathia series. Br J Plast Surg 1991; 44: 386-389. [ Links ]
12. Hide T, Hatakeyama J, Kimura-Yoshida C, Tian E, Takeda N, Ushio Y, et al. Genetic modifiers of otocephalic phenotypes in Otx2 heterozygous mutant mice. Development 2002; 129: 4347-4357.. [ Links ]
13. Schiffer C, Tariverdian G, Schiesser M, Thomas MC, Sergi C. Agnathia-otocephaly complex: report of three cases with involvement of two different Carnegie stages. Am J Med Genet 2002; 112: 203-208.. [ Links ]
14. Martínez-Frías ML, Bermejo E. Frequency and trends of congenital defects in Spain: usefulness and significance of different frequencies. Med Clin (Barcelona) 1999; 113: 459-462[ [ Links ]STANDARDIZEDENDPARAG]
15. Cohen M. The Sonic Hedgehog Signaling Pathway. En: Inborn errors of development. New York: Oxford University Press; 2004. p. 210-228. [ Links ]

