










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombia Médica
On-line version ISSN 1657-9534
Colomb. Med. vol.56 no.1 Cali Jan./Mar. 2008
Insulina cerebral
Andrés Jagua1, Rafael Alejandro Marín, MD2, Luis Alexander Granados, MD3, Vladimir Ávila, MD4
1. Estudiante, Quinto semestre de Medicina, Universidad Nacional de Colombia, Bogotá, Colombia.
e-mail: ajaguag@unal.edu.co
2. Médico Cirujano, Universidad Nacional de Colombia, Bogotá, Colombia. e-mail: neuromarin@hotmail.com
3. Residente, Tercer año de Neurología Clínica, Universidad Nacional de Colombia, Bogotá, Colombia.
e-mail: lagranadosp@unal.edu.co
4. Docente Auxiliar, Universidad Nacional y Universidad Antonio Nariño, Bogotá, Colombia.
e-mail: vlademeco@hotmail.com
Recibido para publicación enero 15, 2007 Aceptado para publicación enero 31, 2008
RESUMEN
La insulina es una hormona con efectos sobre el metabolismo y crecimiento normal de muchas células del cuerpo. En las últimas décadas se han descubierto, además, sus efectos sobre funciones del sistema nervioso central: modulación del ciclo apetito-saciedad, función reproductiva, liberación de neurotransmisores, supervivencia neuronal y plasticidad sináptica. Las evidencias obtenidas desde modelos animales y hallazgos neuropatológicos han permitido entender parte de los mecanismos que asocian a la señal de la insulina con enfermedades neurodegenerativas como la enfermedad de Alzheimer. En este artículo se revisarán las acciones de la insulina sobre el hipotálamo, la supervivencia neuronal, la plasticidad sináptica y por último, las implicaciones de estos conocimientos en la comprensión de procesos degenerativos del sistema nervioso central como la enfermedad de Alzheimer.
Palabras clave: Insulina; Receptor de insulina; Hipotálamo; Supervivencia celular; Enfermedad de Alzheimer; Aprendizaje y memoria.
Brain insulin
SUMMARY
Insulin acts on metabolisms and normal growth. In the past decades, its effects on central nervous system functions, such as: appetite-satiety cycle modulation, reproductive function, neurotransmitter release, neuronal survival and synaptic plasticity have been discovered. Evidences got from animal models and neuropathologic findings, have elucidated a fraction of the mechanism that associates insulin signal with neurodegenerative syndromes, like Alzheimer disease. Here, insulin action on hypothalamus, neuron survival, synaptic plasticity and implications of this knowledge on understanding degenerative process of central nervous system particularly Alzheimer disease, will be reviewed.
Keywords: Insulin; Insulin receptor; Hypothalamus; Cell survival; Alzheimer disease; Learning and memory.
Históricamente, el cerebro se ha caracterizado como un tejido insensible a la insulina. Sin embargo, ciertos hallazgos obtenidos en los últimos treinta años sugieren que la insulina es esencial para las funciones del sistema nervioso central1.
Actualmente se ha establecido que la diabetes mellitus es un factor de riesgo para el desarrollo de la enfermedad de Alzheimer (EA); estudios basados en muestras de patología de cerebros de pacientes muertos con EA revelan productos glicosilados en los agregados de proteínas β-amiloides y el síndrome de resistencia a la insulina se asocia con la EA de manera independiente del fenotipo APOE42. La EA podría ser en parte resultado de un defecto en la señal de la insulina sobre las neuronas, lo cual ha llevado a especular sobre si la EA podría considerarse como otra clase de diabetes (¿diabetes tipo 3?)3.
En primer lugar se revisarán las fuentes y las vías de señalización activadas por la insulina cerebral. Después, se analizará el papel de la insulina en los procesos de plasticidad sináptica, aprendizaje y memoria. Por último, se relacionarán estos hallazgos con las enfermedades neurodegenerativas particularmente con la EA.
LAS FUENTES DE LA INSULINA CEREBRAL
La procedencia de la insulina cerebral es aun tema de controversia. El origen de la insulina cerebral es motivo de discusiones; si bien lo que más se acepta en la actualidad es que existe un sistema de transporte de insulina a través de la barrera hematoencefálica (BHE), algunas evidencias en animales hacen pensar que podría haber expresión de insulina en las neuronas.
La insulina producida por las células del páncreas atraviesa la BHE por un sistema de transporte mediado por el receptor de la insulina (transcitosis). En condiciones normales, los niveles de insulina cerebral se correlacionan con las concentraciones en la circulación periférica4, 5.
Los estudios con insulina marcada demuestran que atraviesa la BHE; bien podría ser, que toda la insulina cerebral proceda del páncreas y su trasporte se dé de una manera saturable (pues está limitado por la disponibilidad de receptores de insulina para su transporte). La BHE es dinámica, se adapta a los niveles periféricos de insulina: durante el estado de ayuno decrecen los niveles cerebrales de insulina; más aún, no todas las regiones de la barrera son igualmente permeables: a nivel del puente y de la médula oblonga se encuentran los mayores niveles de permeabilidad; la permeabilidad a nivel del hipotálamo y del hipocampo también es importante aunque considerablemente menor que la de las áreas antes citadas; en las regiones del cuerpo estriado, cerebelo, corteza frontal y corteza parietal se presentan también niveles de permeabilidad; y no se registra permeabilidad en regiones del mesencéfalo, tálamo y corteza occipital6. La expresión de receptores de insulina en los distintos sitios de la BHE determina el grado de permeabilidad, aunque las diferencias de expresión de estos receptores no se han explorado del todo7.
Sin embargo, algunos investigadores demostraron la expresión de mRNA del gen de la insulina, sin que esto signifique que el mRNA continúe el proceso de transducción5.
El receptor de la insulina se expresa en algunas regiones del encéfalo. El receptor de insulina (RI) es una glicoproteína de 300 a 400 kDa, conformada por dos cadenas alfa idénticas que se ubican en la región extracelular y dos subunidades beta que terminan dentro del citosol; las cadenas beta poseen actividad tirosina quinasa intrínseca8.
El RI se encuentra en mayor concentración en las neuronas cuando se las compara con las células gliales. Su peso molecular es similar al peso del receptor ubicado en otros tejidos, conservándose ampliamente la subunidad beta del receptor. Se demostró un patrón de glicosilación diferente para el receptor expresado en el tejido nervioso, con algunas variaciones en la región antigénica del sitio de unión a la insulina que no puede atribuirse a la disposición del ácido siálico y posibles variaciones en la secuencia de aminoácidos que aún están sin explorar y que podrían modificar la capacidad del receptor para unirse a la insulina9.
El RI se encuentra diferencialmente en las estructuras encefálicas y a densidades distintas; se ha demostrado su expresión a nivel del bulbo olfatorio, hipotálamo, glándula pituitaria, plexo coroideo, núcleos talámicos, corteza piriforme, formación hipocampal, núcleos amigdalinos, corteza prefrontal y cerebelo (las más importantes concentraciones del RI se encuentran a nivel del bulbo olfatorio, hipotálamo, hipocampo y cerebelo)5, 10, 11.
INSULINA Y SUPERVIVENCIA NEURONAL
Una vez que la insulina atraviesa la BHE (o ha sido producida por algunos grupos de neuronas) está disponible para unirse a su receptor. Esta unión activa a la subunidad b del receptor, se autofosforila y ahora posee la capacidad de fosforilar otras proteínas12.
El sustrato del RI-1 (IRS-1) y el sustrato del RI-p58/p53 se expresan en los mismos sitios que el RI13, 14. Estos IRS pueden activar diversas vías, entre ellas, la vía mediada por la fosfatidil inositol 3 quinasa (PI3K) que es la integradora de las señales de la insulina cerebral. La PI3K es un heterodímero con una subunidad catalítica y una reguladora (p110 de cuatro isoformas y p55g, p8a, p85b) que une al IRS a través de sus dominios SH2; activa, la PI3K convierte el fosfatidilinositol 4, 5 bifosfato (PIP2) en fosfatidilinositol 3, 4, 5-trifosfato (PIP3). El PIP3 se une a la proteína quinasa B (PKB/Akt) y otra proteína quinasa, la PDK1 la fosforila y activa (Thr308 para la isoforma PKBb, Thr309 en la isoforma PKBb, Thr305 para la PKBg)8.
La PKB fosforila en Ser136 a la BAD y evita que se una a la BCL-XL y se inicie una cascada proapotótica; la PKB puede también inhibir la ruta de las caspasas (proteínas apoptóticas), promueve la expresión de inhibidores de estas proteínas (FLIP), reduce la expresión de las proteínas relacionadas con el Fas-ligando e inactiva a la ASK1 (quinasa reguladora de la señal de la apoptosis-1) que activa las proteínas relacionadas con la apoptosis15, 16.
Los miembros de la familia de factores de trascripción FoxO, cuya expresión se demostró en las neuronas, poseen motivos de unión a la PKB y son un blanco de control del crecimiento y supervivencia neuronal17.
La quinasa de la glicógeno sintasa 3 (GSK3) fosforila en la neurona proteínas asociadas con los microtúbulos (como las proteínas Tau); precisamente, la hiperfosforilación de las Tau se asocia con procesos neurodegenerativos y la insulina por la vía PI3K-PKB regula su nivel de fosforilación18.
La insulina puede activar también a nivel de la neurona, la vía de las MAP quinasas (proteínas activadas por mitógeno). La Grb2 contiene dominios SH2 que le permiten interactuar con el IRS y, además, por su dominio SH3 une a una región rica en prolina de la Sos, formando un complejo. Sos unido a Grb2 puede ahora catalizar el intercambio de GDP (difosfato de guanosina) por GTP (trifosfato de guanosina) sobre RAS. RAS, que se ha activado por su unión con el GTP inicia la activación de la Raf (una serina treonina quinasa) que fosforila y activa a la MEK (treonina-tirosina quinasa) que finalmente fosforila y activa a la ERK. La ERK fosforila a otras proteínas en el citosol con blancos nucleares y posee directamente múltiples objetivos nucleares que modulan los procesos de transcripción de los genes (Elk-1, SRF, jun/fos)8, 19, 20. Todas estas evidencias son consistentes con los modelos que hablan de la insulina como factor neurotrófico.
Xu et al.20 comprobaron que administrar etanol a ratas hembras durante la gestación resulta en hipoplasia del cerebelo en desarrollo, por un mecanismo que inhibe la acción neuroprotectora de la insulina. La alteración de su señal comienza a nivel del RI (por una disminución en su expresión) e incluye a los mediadores cascada abajo (descenso del IRS-1, Akt, aumento del BAD)21.
En otro estudio Hui et al.21 describen una nueva vía inducida por la insulina luego de un episodio de isquemia cerebral y que resulta en procesos neuroprotectivos. Diseñaron un protocolo en el que se sometía a ratas (Sprague-Dawley) a quince minutos de isquemia cerebral por oclusión de la carótida. Las ratas que recibieron insulina veinte minutos antes del episodio de isquemia, mostraron una mejor evolución comparadas con el grupo de control. La insulina al actuar activa su receptor que a través de la vía IRS/PI3K/Akt dirige la inhibición de la JNK (c-jun quinasa N-terminal) relacionada con la activación de la BCL-2 (por fosforilación en Ser87) y la continuación de una cascada apóptótica22.
LA INSULINA EN EL HIPOTÁLAMO
La insulina obra de manera conjunta con la leptina en la regulación del ciclo apetito-saciedad; es un agente anorexigénico e induce la disminución del peso corporal23. El IRS-2 modula el comportamiento alimenticio y reproductor; modelos de ratones IRS-2-/- (knockout para el IRS-2) dan como resultado ratones hiperfágicos y obesos como consecuencia de la defectuosa señal de la insulina4, 24. Una población de neuronas en el núcleo arcuato expresa POMC (propiomelanocortina), precursor delα-MSH (hormona estimulante de los melanocitos) que interactúa con sus receptores en otras neuronas hipotalámicas e induce la sensación de apetito; a este sistema lo modulan la insulina y la leptina25.
Los niveles de expresión de mRNA del neuropéptido Y (asociado con la sensación de apetito) en el núcleo arcuato se ven afectados como resultado de la señal de la insulina mediante un aumento de la liberación de neurotransmisores inhibitorios (sistema dependiente de GABAA) o por la disminución de los excitatorios, sin embargo el proceso no se comprende del todo26, 27. Se ha demostrado también que la insulina activa canales de K (potasio) sensibles a ATP en las neuronas del hipotálamo vía PI3K, y dirige la reducción de la sensación de apetito y la baja del peso corporal en ratones no obesos28.
La administración intranasal de insulina durante períodos prolongados, hace que se reduzca el peso corporal en hombres con disminución del contenido de grasa, pero en las mujeres parece no haber pérdida de peso debida a la ganancia de agua extracelular29.
El control del equilibrio energético está profundamente ligado a la función reproductiva. Las células productoras de GnRH expresan el receptor de insulina; ratones knockout para el RI muestran alteraciones en la espermatogénesis y en la maduración del folículo ovárico30. La insulina podría ser un elemento clave dentro de una red hormonal que informa al encéfalo sobre el estado general del cuerpo (estado nutricional, relación talla-peso) para que éste decida la activación o no de la función reproductiva a través de la liberación de la GnRH.
La insulina también podría regular la función hipotalámica, pues contribuye al mantenimiento de los procesos de la memoria, en particular, en el consumo de comida, y al parecer, está relacionada con los proceso de aprendizaje del contenido calórico de los alimentos31, 32. Los RI expresados en el hipocampo podrían hacer parte de una red de comunicación en la que el hipotálamo suministra informe sobre el equilibrio energético del cuerpo; o bien la insulina que actúa directamente sobre los RI del hipocampo podría codificar datos sobre los contenidos energéticos de los alimentos que se consumen, y que se procesarían y emplearían después ante una nueva ingesta de comida de manera que se activen circuitos para aumentar o disminuir la sensación de apetito (por comparación del contenido calórico aprendido y el consumido).
INSULINA Y PLASTICIDAD SINÁPTICA
La expresión de receptores de insulina en el hipocampo ha llamado la atención de los investigadores por sus posibles papeles sobre el aprendizaje y la memoria.
Tanto la memoria declarativa como la no declarativa se relacionan con la formación del hipocampo y se codifican mediante un alfabeto molecular complejo y redes neuronales extensas33.
Zhao et al.9 demostraron un incremento en la concentración de mRNA (RNA mensajero) del RI en el hipocampo de ratas (cepa Wistar) luego de entrenarlas en laberintos de agua; mostraron además, en pruebas in vitro, que la actividad tirosina quinasa del receptor era mucho mayor en los ratones entrenados. El hipocampo es un área de gran importancia en los procesos de aprendizaje y estas evidencias sugieren un papel para la insulina sobre los procesos de formación de la memoria10.
Estudios en moluscos marinos del género Applysia muestran que la insulina causa un aumento rápido en el nivel de calcio intracelular en ausencia de potenciales de acción. Este calcio proviene de un conjunto (pool) independiente al liberado por acción del IP3 (trifosfato de inositol) ya que la administración de antagonistas del receptor de IP3 en el retículo endoplásmico no altera los aumentos de calcio debidos a la señal de la insulina. El aumento del calcio permite entre otras actividades la liberación de vesículas sinápticas (también ocurre la activación de proteínas relacionadas con la regulación de la trascripción genética y la activación de otras proteínas como la CAMKII)34.
Los modelos en ratas Wistar muestran detalles interesantes. Un estudio dirigido por Babri et al.34 indica que la insulina podría relacionarse con los procesos de consolidación de la memoria. En otro estudio de Dou et al.35 con ratas diabéticas (por administración de estreptozotocina) sometidas a tareas de memoria espacial, se demuestra que si bien la adquisición de memoria en estas ratas es normal, si ocurre una alteración de los procesos de consolidación de la memoria a largo plazo; el aumento en la actividad del RI, de la cantidad de Shc unida a la membrana (estado activo), de la actividad de la ERK 1/2 y de la actividad de Akt se relacionan con la potenciación de la memoria a largo plazo36.
Los estudios en seres humanos arrojan también resultados que refuerzan la idea de las acciones de la insulina sobre los procesos de la memoria. En un estudio donde se sometió a condiciones de hiperinsulinemia a un grupo de individuos con EA, se observó que las tareas de memoria declarativa mejoraban considerablemente comparados con el grupo control, de manera selectiva e independiente de los niveles de glucosa en la sangre37. Otros estudios comunican también mejoras en los procesos de memoria declarativa29. De igual forma, la insulina por vía intranasal mejora la atención selectiva y facilita codificar informes importantes al modular la recaptación de noradrenalina en el hipocampo38.
La insulina puede incrementar el número de los receptores de GABAA (receptores para ácidoγ-aminobutírico) disponibles en la membrana postsináptica sin que su síntesis esté comprometida, es decir, la señal de la insulina induce la activación de sistemas complejos de señalización intracelular como el sistema de las ubiquitinas para reclutar y organizar el tráfico de receptores39; inhibir la actividad tirosina quinasa de la subunidad b del RI resulta en la interrupción del aumento de receptores de GABAA en la membrana postsináptica cerca de los sitios de liberación de neurotransmisores desde la membrana presináptica40. Los cambios en la efectividad sináptica que se consideran sustrato molecular de los procesos de la memoria, se deben en parte al aumento de la disponibilidad de receptores de las sinapsis33.
El RI se expresa densamente en las sinapsis y en algunas regiones de las dendritas en las neuronas de muchas regiones del encéfalo13; y el RI, el IRS y la actividad de tirosina quinasa del RI se han podido caracterizar en las mismas regiones y demostrar su co-localización14. La insulina fosforila (vía RI) a la PDK1, Akt, mTor (blanco mamífero de rapamicina) y 4E-BP1. El 4E-BP1 actúa como un represor de la transcripción que al ser fosforilado cesa su actividad represora, y se convierte en un punto de modulación de la expresión de genes necesarios para la generación de memoria; así, por lo menos a nivel del hipocampo, la insulina por la ruta PI3K/Akt/mTor modula la disponibilidad de proteínas del sinaptosoma como la PSD-95 (proteína de densidad postsináptica 95) que eleva la sensibilidad del NMDA-R (receptor de glutamato tipo N-metil-D-aspartato)41. Por otra parte, las señales que envía la insulina (ERK, PI3K, aumento del calcio intracelular y la activación de sus diversos blancos como la CAMKII, proteínas reguladoras del tráfico vesicular y el CREB) a nivel del hipocampo son comunes con los procesos moleculares que ocurren durante la formación de la memoria y su potenciación largo plazo42.
La activación del receptor de glutamato tipo NMDA parece aumentar la disponibilidad del IRSp53 en las espinas dendríticas. La administración de antagonistas de la PKC (activada por el NMDA-R) conduce al descenso de su concentración. El mecanismo de reclutamiento del IRSp53 se relaciona con la actina y bien podría actuar en procesos de maduración sináptica43.
La magnitud de los hallazgos abre puertas esperanzadoras para el tratamiento de los trastornos cognitivos. Sin embargo, los mecanismos son complejos y aún poco comprendidos; se requieren más estudios para establecer la totalidad de los efectos de la insulina sobre la función cognitiva44. Los principales sucesos moleculares ya explicados se resumen en la Figura 1.
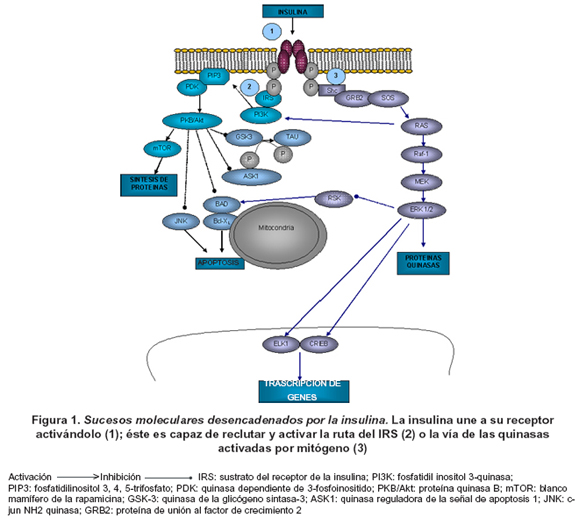
Los defectos de la acción de la insulina cerebral pueden provocar el desarrollo de procesos neurodegenerativos
La insulina parece cumplir un papel importante en el funcionamiento normal de las neuronas. Las anormalidades en sus actividades se han relacionado con la EA3, el mal de Parkinson45 y la depresión46. En lo que resta del artículo se analizarán las implicaciones para la EA.
La EA muestra un patrón degenerativo asociado con deficiencias de la función cognitiva. La EA es la demencia mas común entre los adultos mayores; cerca de cuatro millones de personas en los Estados Unidos tienen EA y su costo aproximado es de cien billones de dólares anuales47. La incidencia de la enfermedad aumentará con los años y los esfuerzos dirigidos a la búsqueda de nuevos tratamientos se justifican ampliamente48.
Se han descrito anormalidades estructurales a nivel del hipocampo, corteza entorrinal, amígdala, núcleos basales, núcleo anterior del tálamo y neocorteza. Pueden distinguirse agregados compuestos sobre todo de proteína β-amiloide (4kDa) que resulta del clivaje de la proteína precursora de amiloide (PPA). La PPA es miembro de una familia de proteínas relacionadas también con la enfermedad (APLP1 y APLP2) codificadas por genes ubicados en el brazo largo del cromosoma 2149. La presenilina 1 (PSNE1) y 2 (PSNE2), la APOE4 (apolipoproteína E4), junto con la PPA parecen conferir susceptibilidad genética para la EA50.
A nivel de la neurona se identifican «ovillos» de proteína TAU hiperfosforilada. La TAU es una proteína de 50 a 65 kDa asociada con los microtúbulos que cumple importantes funciones en la organización del citoesqueleto neuronal y el tráfico vesicular51. También se aprecian distrofia neurítica, pérdida de sinapsis y muerte neuronal52, 53.
Insulina y EA. ¿es la EA una diabetes mellitus tipo 3? Cuando se comparan individuos diabéticos con personas sanas se aprecian las alteraciones cognitivas con mucha más ocurrencia en los enfermos54.
En general la resistencia a la insulina se asocia con la EA independientemente del fenotipo APOE4 (que confiere susceptibilidad a la enfermedad y se asocia con 50% de los casos de la EA, este hecho, refuerza la necesidad de buscar otras causas a la enfermedad); en los agregados β-amiloides se depositan productos glicosilados. La EA podría desarrollarse como una resistencia neuronal a la acción de la insulina2.
La insulina aumenta la captación de lipoproteínas mediada por APOE en el tejido nervioso a través de la proteína relacionada con el receptor de lipoproteína (LRP), la LRP degrada la PPA. La insulina también afecta los niveles de acetilcolina en el hipocampo y reduce la fosforilación de las proteínas TAU. Se han informado algunas diferencias de las acciones mediadas por la insulina entre los géneros55.
Estudios de tejido cerebral post-mortem muestran una disminución considerable en la densidad de los RI en personas con diagnóstico de EA, sumado a indicios de la disminución de la actividad tirosina quinasa del receptor sin que las concentraciones para otros factores neurotróficos, IGF-I e IGF II se encuentren alterados revelando un mecanismo específico para la insulina56.
La hiperinsulinemia periférica crónica causa una regulación hacia abajo -downregulation- del transporte de insulina a través de la BHE lo cual explicaría los niveles disminuidos de insulina encontrados en los cerebros de los pacientes con EA. Como resultado hay un descenso en la señal de la insulina hacia las neuronas que genera desequilibrio en liberar neurotransmisores, una señal de supervivencia neuronal defectuosa y mala regulación de las enzimas degradantes de insulina57, 58.
Las enzimas degradantes de insulina (EDI) son Zn2+-metaloproteasas de 113 kDa, altamente conservadas a lo largo del proceso evolutivo, presentes sobre todo en el medio extracelular y peroxisomas. Los sustratos para las EDI son la cadena β de la insulina, los péptidos β-amiloides (AB), la amilina y el glucagón. Las EDI degradan AB (variantes 1 a 40) con un pH óptimo de 4 a 5.559. Por tanto las EDI están directamente comprometidas en el aclaramientode los agregados de β-amiloides60 y, en neuronas en cultivo, son capaces de disminuir la toxicidad debida a los AB61.
Las EDI reconocen una secuencia homóloga en la insulina y los AB (residuos 16 a 25 de los AB y 21 a 30 de la cadena B de la insulina) de manera mucho más ávida por la insulina que por los AB62.
Los niveles de EDI en el hipocampo descienden con la edad lo que explicaría su susceptibilidad a la acumulación de AB de esta región comparada con el cerebelo que conserva altos niveles de EDI63. Algunas variantes de EDI (EDI_7, EDI_9, EDI_14) parecen conferir susceptibilidad genética adicional a la acumulación de los AB64-66; una actividad disminuida de las EDI se correlaciona con un alza en los niveles de AB67.
La resistencia a la insulina promueve la acumulación de AB por descenso de la actividad de las EDI. La insulina activa a las EDI y los bajos niveles de insulina informados en los pacientes con EA pueden explicar una activación menor de las EDI y mayor acumulación de los AB. Los AB impiden la acción de la insulina, pues forman un círculo vicioso que termina con la producción de más AB68. El aumento de los AB es capaz también de aumentar la concentración de acetilcolinesterasa consistente con los niveles bajos de acetilcolina típicos de la EA55.
Por otra parte los modelos en ratones con EA muestran que ante el descenso de la concentración de insulina cerebral sus niveles de GSK-3 aumentan, y consigo, los niveles de proteína TAU fosforilada; esto conduce a desórdenes del tráfico vesicular y lleva a muerte neuronal55. La inyección de inhibidores específicos de la PI3K y la PKC resulta en un alza en la actividad de la GSK-3 un día después; se encuentra un aumento en la concentración de TAU fosforilada en Ser 199/202 que eleva así la susceptibilidad para formar ovillos de proteínas TAU69.
El debate sobre si la EA se puede clasificar como una clase de diabetes está abierto. Aún se desconocen muchos de los mecanismos moleculares que relacionan la EA con la resistencia a la insulina, sin embargo, son interesantes las perspectivas que se abren a la luz de los conocimientos actuales.
HORIZONTES FUTUROS
Si bien queda mucho por descubrir, los conocimientos actuales acerca de la acción de la insulina sobre el sistema nervioso central permiten abrir nuevos horizontes en el tratamiento de la obesidad, las enfermedades neurodegenerativas y ciertas deficiencias cognitivas.
El papel modulador de la insulina en el ciclo apetito-saciedad es un punto susceptible para el desarrollo de estrategias basadas en su acción sobre el hipotálamo tendiente al tratamiento de los desórdenes de la alimentación.
La protección que confiere la insulina sobre las neuronas se podría emplear en el tratamiento de pacientes que han sufrido isquemia cerebral para mitigar los daños estructurales y neuronales.
Las mejoras de las tareas cognitivas observadas incluso en pacientes con Alzheimer han llevado a pensar su uso para mitigar las deficiencias en las funciones ejecutivas que se presentan en la enfermedad. Asimismo, se ha planteado la posibilidad de potenciar la acción de las enzimas degradantes de insulina (bien sea por terapia génica o por inducción enzimática) como mecanismo para reducir los niveles de AB.
Recientemente Burns et al.70 presentan los resultados de un estudio que incluyó a 31 individuos sin demencia y 31 con EA en estadío temprano. Ellos informan que los niveles de insulina se relacionan de manera directa y proporcional con el volumen del cerebro y la presencia o no de atrofia hipocampal. También encuentran que los niveles altos de insulina se asocian con un mejor comportamiento cognitivo global70.
Por otra parte Reger et al.71 estudian la administración de insulina por vía intranasal (20 UI) en personas con EA en estadío temprano o con MCI (alteración cognitiva leve) comparándolas con un grupo placebo. El tratamiento con insulina no sólo mejora el perfil cognitivo sino además, modula los niveles de AB postpandrial vía reducción de los niveles de cortisol.
Otra aproximación terapéutica que ha sido propuesto a la luz de los hallazgos en esta revisión es el uso de tiazolindinedionas. En una revisión sistemática de la literatura que realizaron los autores de este artículo (información no publicada) no se encontró ningún beneficio con este tratamiento. La limitación más importante de todos los estudios incluidos es que el manejo está dirigido a individuos con diagnóstico de EA y el tratamiento con estos fármacos podría ser útil sólo en casos pre-clínicos de la entidad.
CONCLUSIÓN
- La insulina actúa como un factor neurotrófico indispensable para la supervivencia neuronal; es un integrador de señales centro-periferia a nivel hipotalámico y parece relacionarse con el aprendizaje del contenido de calorías de los alimentos.
- Los procesos de formación de la memoria se ven también influidos por la insulina aunque aún se desconocen muchos de los mecanismos por los cuales ésta ejerce la modulación.
- Por último, es importante enfatizar las implicaciones de estos avances en la comprensión de enfermedades neurodegenerativas como la EA que pueden derivar en mejores tratamientos y en general un mejor pronóstico para quien sufre la enfermedad.
AGRADECIMIENTOS
Agradecemos a nuestros compañeros Julián David Jaimes, Manuel Fernando Pineda y Jairo Andrés Virviescas la lectura del manuscrito.
REFERENCIAS
1. Wickelgren I. Tracking insulin to the mind science. Science. 1998; 280: 517-9. [ Links ]
2. Kuusisto J, Koivisto K, Mykkänen L, Helkala EL, Vanhanen M, Hänninen T, et al. Association between Alzheimer's disease independently of apolipoprotein E4 phenotipe: cross sectional population based study. BMJ. 1997; 315: 1045-9. [ Links ]
3. Pilcher H. Alzheimer's disease could be «type 3 diabetes». Lancet Neurol. 2006; 5: 388-9. [ Links ]
4. Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. 2005; 115: 951-8. [ Links ]
5. Van der Heide LP, Ramakers GM, Smidt MP. Insulin signaling in the central nervous system: learning to survive. Prog Neurobiol. 2006; 79: 205-21. [ Links ]
6. Banks WA, Kastin AJ. Differential permeability of the blood-brain barrier to two pancreatic peptides: insulin and amylin. Peptides. 1998; 19: 883-9. [ Links ]
7. Banks WA. The source of cerebral insulin. Eur J Pharmacol. 2004; 19: 5-12. [ Links ]
8. Nelson DL, Cox MM. Molecular mechanisms of signal transduction. En: Lehninger Principles of biochemistry. 4th ed. New York: WH Freeman; 2005. p. 421-79. [ Links ]
9. Roth RA, Morgan DO, Beaudoin J, Sara V. Purification and characterization of the human brain insulin receptor. J Biol Chem. 1986; 261: 3753-7. [ Links ]
10. Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ, et al. Brain insulin receptors and spatial memory. J Biol Chem. 1999; 274: 34893-902. [ Links ]
11. Kubota N, Terauchi Y, Tobe K, Yano W, Suzuki R, Veki K, et al. Insulin receptor substrate 2 plays a crucial role in β cells and the hypothalamus. J Clin Invest. 2004; 114: 917-24. [ Links ]
12. Litwack G, Schmidt TU. Bioquímica de las hormonas I: Hormonas polipeptídicas. En: Devlink TH editor. Barcelona: Editorial Reverté; 2000. p. 839-92. [ Links ]
13. Baskin DG, Schwartz MW, Sipols AJ, D'Alessio DA, Goldstein BJ, White MF. Insulin receptor substrate-1 (IRS-1) expression in rat brain. Endocrinology. 1994; 134: 1952-5. [ Links ]
14. Abbott M, Wells DG, Fallon JR. The insulin receptor tyrosine kinase substrate p58/p53 and the insulin receptor are components of CNS synapses. J Neurosci. 1999; 19: 7300-8. [ Links ]
15. Lawlor MA, Alessi DR. PKB/AKT: a key mediator of cell proliferation, survival and insulin responses? J Cell Sci. 2001; 114: 1142903-10. [ Links ]
16. Jessell TM, Soines JR. The generation and survival of nerve cells. En: Kandel ER, Schwartz JH, Jessel TM editors. Principles of neural sciences. New York: McGraw-Hill; 2000. p. 1041-62. [ Links ]
17. Jacobs FM, Van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, Smidt MP. FoxO6, a novel member of the FoxO class of transcription factors with distinct shauttling dynamics. J Biol Chem. 2003; 278: 35959-67. [ Links ]
18. Hong M, Lee VM. Insulin and insulin-like growth factor-1 regulate Tau phosphorylation in cultured human neurons. J Biol Chem. 1997; 272: 19547-53. [ Links ]
19. Karp G. Señales celulares: comunicación entre células y su ambiente. En: Biología celular y molecular. México: McGraw-Hill Interamericana; 2003. p. 325-88. [ Links ]
20. Alberts B, Bray D, Lewis J, Raff M, Roberts K, Watson JD. Transmisión de señales entre las células. En: Biología molecular de la célula. Barcelona: Ediciones Omega; 1996. p. 771-841. [ Links ]
21. Xu J, Yeon JE, Chang H, Tison G, Chen GJ, Wands J, et al. Ethanol impairs insulin-stimulated neuronal survival in the developing brain: role of PTEN phosphatase. J Biol Chem. 2003; 278: 26929-37. [ Links ]
22. Hui L, Pei DS, Zhang QG, Guan QH, Zhang GY. The neuroprotection of insulin on ischemic brain injury in rat hippocampus through negative regulation of JNK signaling pathway by PI3K/Akt activation. Brain Res. 2005; 1052: 1-9. [ Links ]
23. Air EL, Strowski MZ, Benoit SC, Conarello SL, Salituro M, Guan XM, et al. Small molecule insulin mimetic reduces food intake and body weight and prevents development of obesity. Nat Med. 2002; 8: 179-83. [ Links ]
24. Masaki T, Chiba S, Noguchi H, Yasuda T, Tobe K, Suzuki R, et al. Obesity in insulin receptor substrate-2 deficient mice: disrupted control of arcuate nucleus neuropeptides. Obesity Res. 2004; 12: 878-85. [ Links ]
25. Benoit SC, Air EL, Coolen LM, Strauss R, Jackman A, Clegg DJ, et al. The catabolic action of insulin in the brain is mediated by melanocortins. J Neurosci. 2002; 22: 9046-52. [ Links ]
26. Sato I, Arima H, Ozaki N, Watanabe M, Goto M, Hayashi M, et al. Insulin inhibits neuropeptide and gene expression in the arcuate nucleus through GABAergic systems. J Neurosci. 2005; 25: 8657-64. [ Links ]
27. Kupfermann I, Kandel ER, Iverse N. Motivational and adictive sates. En: Kandel ER, Schwartz JH, Jessel TM editors. Principles of neural sciences. New York: McGraw-Hill; 2000. p. 998-1013. [ Links ]
28. Spanswick D, Smith MA, Mirshamsi S, Routh VH, Ashford ML. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci. 2000; 3: 757-8. [ Links ]
29. Hallschmid M, Benedict C, Schultes B, Fehm H, Born J, Kein W. Intranasal insulin reduces body fat in men but not in women. Diabetes. 2004; 53: 3024-29. [ Links ]
30. Brüning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban DC, et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000; 289: 2122-5. [ Links ]
31. Gerozissis K. Brain insulin: regulation, mechanisms of action and functions. Cel Mol Neurobiol. 2003; 23: 1-25. [ Links ]
32. Gerozissis K, Rouch C, Lemierre S, Nicolaidis S, Orosco M. A potential role of central insulin in learning and memory related to feeding. Cel Mol Neurobiol. 2001; 21: 389-401. [ Links ]
33. Kandel ER. Celular mechanisms of learning and biological basis of individuality. En: Kandel ER, Schwartz JH, Jessel TM editors. Principles of neural sciences. New York: McGraw-Hill; 2000. p. 1247-79. [ Links ]
34. Jonas EA, Knox RJ, Smith TC, Wayne NL, Connor JA, Kaczmarek LK. Regulation by insulin of unique neuronal Ca2+ pool and of neuropeptid secretion. Nature. 1997; 385: 343-6. [ Links ]
35. Babri S, Badie HG, Kahamenei S, Seyedlar MO. Intrahippocampal insulin improves memory in a passive-avoidance task in male Wistar rats. Brain Cogn. 2007; 64: 86-91. [ Links ]
36. Dou JT, Chen M, Dufour F, Alkon DL, Zhao WQ. Insulin receptor signaling in long-term memory consolidation following spatial learning. Learn Mem. 2005; 12: 646-55. [ Links ]
37. Craft S, Asthana S, Newcomer JW, Wilkinson CW, Matos IT, Baker LD, et al. Enhancement of memory in Alzheimer's disease with insulin and somatostatin, but not glucose. Arch Gen Psych. 1999; 56: 1135-40. [ Links ]
38. Kern W, Peters A, FruehwoldSchultes B, Deininger E, Born J, Fehm HL. Improving influence of insulin on cognitive functions in humans. Neuroendocrinology. 2001; 74: 270-80. [ Links ]
39. Haglund K, Dikic I. Ubiquitylation and cell signaling. EMBO J. 2005 5; 24: 3353-59. [ Links ]
40. Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, et al. Recruitment of functional GABAA receptors to postsynaptic domains by insulin. Nature. 1997; 338: 686-90. [ Links ]
41. Lee CC, Huang CC, Wu MY, Hsu KS. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J Biol Chem. 2005; 280: 18543-550. [ Links ]
42. Lynch MA. Long term potentiation and memory. Physiol Rev. 2004; 84: 87-136. [ Links ]
43. Hori K, Yasuda H, Konno D, Maruoka H, Tsumoto T, Sobue K. NMDA receptor-dependent synaptic translocation of insulin receptor substrate p53 via protein kinase C signaling. J Neurosci. 2005; 25: 2670-81. [ Links ]
44. Strachan MW. Insulin and cognitive function in humans: experimental data and therapeutic considerations. Biochem Soc Transc. 2005; 33: 1037-40. [ Links ]
45. Craft S, Watson GS. Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol. 2004; 3: 169-78. [ Links ]
46. Rasgon NL, Kenna HA. Insulin resistance in depressive disorders and Alzheimer's disease: revisiting the missing link hypothesis. Neurobiol Aging. 2005; 265: 5103-7. [ Links ]
47. Mandavilli A. The amyloid code. Nat Med. 2006; 12: 747-51. [ Links ]
48. Hodes RJ. Public Funding for Alzheimer's disease research in the United States. Nat Med. 2006; 12: 770-3. [ Links ]
49. Price DL. Aging of the brain and dementia of the Alzheimer type. En: Kandel ER, Schwartz JH, Jessel TM editores. Principles of neural sciences. New York: McGraw-Hill; 2000. p. 1149-68. [ Links ]
50. Shinkai Y, Yoshimura M, Morishima-Kawashima M, Ito Y, Shimada H, Yanagisawa K, et al. Amyloid beta-protein deposition in the leptomeninges and cerebral cortex. Ann Neurol. 1997; 42: 899-908. [ Links ]
51. Karp G. Citoesqueleto y motilidad celular. En: Biología celular y molecular. México: McGraw-Hill Interamericana; 2003. p. 325-88. [ Links ]
52. Cole G. A transgenic triple scores a home run. Nat Med. 2006; 12: 762-3. [ Links ]
53. Sinclair AJ, Girling AJ, Bayer AJ. Cognitive dysfunction in older subjects with diabetes mellitus: impact on diabetes self-management and use of care services all walles research into elderly (Aware) study. Diab Res Clin Pract. 2000; 50: 203-12. [ Links ]
54. Qui WQ, Folstein MF. Insulin, insulin-degrading enzyme and amyloid-β peptide in Alzheimer's disease: review and hypothesis. Neurobiol Aging. 2006; 27: 190-8. [ Links ]
55. Craft S, Asthana S, Schellenberg G, Cherrier M, Baker LD, Newcomer J, et al. Insulin metabolism in Alzheimer's disease differs according to apolipoprotein E genotype and gender. Neuroendocrinology. 1999; 70: 146-52. [ Links ]
56. Frölich L, Blum-Degen D, Bernstein HG, Engelsberger S, Hurnrich J, Laufer S, et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer‘s disease. J Neural Transm. 1998; 105: 423-38. [ Links ]
57. Craft S. Insulin resistance syndrome and Alzheimer's disease: age-and obesity-related effects on memory, amyloid and inflammation. Neurobiol Aging. 2005; 26 Supl: 65-9. [ Links ]
58. Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in the Alzheimer‘s disease-is this type 3 diabetes? J Alzheimers Dis. 2005; 7: 63-80. [ Links ]
59. McDermott JR, Gibson AM. Degradation of Alzheimer's β-amyloid proteinby human and rat brain peptidases: involvement of insulin-degrading enzyme. Neurochem Res. 1997; 22: 49-56. [ Links ]
60.Shen Y, Juachimiak A, Rosner MR, Tang WJ. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature. 2006; 443: 870-4. [ Links ]
61. Vekrellis K, Ye Z, Qiu WQ, Walsh D, Hartley D, Chesneau V, et al. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J Neurosci. 2000; 20: 1657-65. [ Links ]
62. Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R. Alzheimer's β-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci. 2002; 22: RC221. [ Links ]
63. Caccamo A, Oddo S, Sugarman MC, Akbari Y, Laferla FM. Age-and region-dependent alterations in Ab-degrading enzymes: implications for Ab-induced disorders. Neurobiol Aging. 2005; 26: 645-54. [ Links ]
64. Blomqvist ME, Chalmers K, Andreasen N, Bogdanovic N, Wilcock GK, Cairns NJ, et al. Sequence variants of IDE are associated with the extend of β-amyloid deposition in the Alzheimer's disease. Neurobiol Aging. 2005; 26: 795-802. [ Links ]
65. Mueller JC, Riemenschneider M, Schoepfer-Wendels A, Gohlke H, Konta L, Friedrich P, et al. Weak independent association signals between IDE polymorphisms, Alzheimer's disease and cognitive measures. Neurobiol Aging. 2007; 28: 727-34. [ Links ]
66. Bjork BF, Katzov H, Kehoe P, Fratiglioni L, Winblad B, Prince JA, et al. Positive association between risk for late-onset Alzheimer's disease and genetic variations in IDE. Neurobiol Aging. 2007; 28: 1374-80. [ Links ]
67. Zhao Z, Xiang Z, Haroutunian V, Buxbaum J, Stetka B, Pasinetti GM. Insulin degrading enzyme activity selectively decreases in the hippocampal formation of cases at high risk to develop Alzheimer's disease. Neurobiol Aging. 2007; 28: 824-30. [ Links ]
68. Zhao L, Teter B, Morihara T, Lim GP, Ambergaokar SS, Ubeda OJ, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J Neurosci. 2004; 24: 11120-6. [ Links ]
69. Liu SJ, Zhang AH, Li HL, Wang Q, Deng HM, Netzer WJ, et al. Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. J Neurochem. 2003; 87: 1333-44. [ Links ]
70. Burns JM, Donnelly JE, Anderson HS, Mayo MS, Spencer-Gardner L, Thomas G, et al. Peripheral insulin and brain structure in early alzheimer disease. Neurology. 2007; 69: 1094-104. [ Links ]
71. Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B, et al. Intranasal insulin improves cognition and modulates β-amyloid in early AD. Neurology. 2008; 70: 440-8. [ Links ]
LISTA DE ABREVIATURAS
EA: Enfermedad de Alzheimer; APOE4: Apolipoproteína E4; BHE: Barrera hemato-encefálica; RI: Receptor de insulina; kDa: Kilodaltones; IRS: sustrato del receptor de la insulina; PI3K: Fosfatidil inositol 3 quinasa; PIP2: Fosfatidil inositol 4, 5 bifosfato; PIP3: Fosfatidil inositol 3, 4, 5 trifosfato; PKB/akt: Proteína quinasa B; PDK1: Quinasa dependiente de 3 fosfoinositido; ASK1: Quinasa reguladora de la señal de la apoptosis 1; GSK3: Quinasa de la glicógeno sintasa 3; MAP: Proteína activada por mitógenos; Grb2: Proteína de unión al factor de crecimiento 2; GDP: difosfato de guanosina; GTP: Trifosfato de guanosina; Elk1: precursor del receptor efrina 1; SRF: Factor de respuesta al suero; FLIP: Proteína inhibidora de la enzima convertidora de Interleuquina 1 β similar al dominio de muerte asociado a FAS; POMC: Propiomelanocortina; α-MSH: Hormona estimulante de los melanocitos; GABA: ácido gamma-aminobutírico; GnRH: Hormona liberadora de gonadotropinas; mRNA: Ácido ribonucleico mensajero; IP3: Trifosfato de inositol; CAMKII: calcio-calmodulina quinasa II; mTOR: blanco mamífero de rapamicina; 4E-BP1: proteína de unión al factor de iniciación eucariota 4E-1; PSD-95: Proteína de densidad postsináptica 95; NMDAR: receptor de glutamato tipo N-Metil-D aspartatO; CREB: proteína de unión al elemento de respuesta al AMPc; IRSp53: sustrato del receptor de insulina p53; PSNE1/2: presenilinas ½; LRP: receptor de lipoproteína; IGF-I: Factor de crecimiento similar a la insulina; ED: enzimas degradantes de insulina; AB: amiloide β.

