










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombia Médica
On-line version ISSN 1657-9534
Colomb. Med. vol.39 suppl.2 Cali June 2008
Detección de portadoras de distrofia muscular de Duchenne en familias colombianas mediante análisis de microsatélites1
Dora Fonseca, MSc2, Claudia Tamar Silva, MSc2, Heidi Mateus, MSc2
1. Proyecto financiado por Colciencias.
2. Profesora Principal, Unidad de Genética, Facultad de Medicina, Universidad del Rosario, Bogotá, Colombia. e-mail: dfonseca@urosario.edu.co ctsilva@urosario.edu.co hmateus@urosario.edu.co
Recibido para publicación junio 25, 2007 Aceptado para publicación abril 18, 2008
RESUMEN
Introducción: Las distrofias musculares de Duchenne y Becker son enfermedades recesivas ligadas al cromosoma X; la identificación de portadoras se puede hacer por métodos directos cuando se ha identificado la mutación, o por indirectos como el análisis de haplotipos.
Objetivo: Se busca establecer mediante análisis de STRs y construcción de haplotipos el estado de portadora o no portadora en 37 familias con afectados por DMD/DMB.
Metodología: Se estudiaron 174 personas mediante el análisis de 10 STRs intra y extragénicos del gen de la distrofina y la construcción de haplotipos para la identificación del ligado a la mutación.
Resultados: Con la metodología mencionada se logró determinar el estado de portadora en 89.2% de las mujeres participantes, de las cuales 65.7% eran portadoras y 23.5% no portadoras.
Conclusiones: El análisis indirecto mediante construcción de haplotipos permitió establecer el estado de portadora en una gran proporción de la población analizada de mujeres y permitió brindar un adecuado asesoramiento genético.
Palabras clave: Duchenne; Diagnóstico; Portador; Microsatélites; Colombia.
Carrier detection of Duchenne muscular dystrophy in Colombian families by microsatellite analysis
SUMMARY
Introduction: The muscular dystrophies of Duchenne and Becker are X-linked recessive neuromuscular disorders; the carrier testing protocols include mutation detection or linkage analysis.
Objective: The aim of this investigation was to use the segregation analysis of STR loci to determine the carrier status in 37 families with DMD/DMB.
Methods: From 37 families 174 individuals were studied through segregation of 10 intra and extragenic short tandem repeats (STR) in the members of the family.
Results: The carrier status of 89.2% women of the tested group could be assigned by linkage analysis, 65.7% carriers and 23.5% non-carriers
Conclusions: Linkage analysis was proven to be a powerful tool for the carrier detection in DMD/BMD and should be taken into account in genetic counselling practice.
Keywords: Duchenne; Carrier; Diagnostic; Colombia; STR.
Las distrofias musculares de Duchenne y Becker (DMD/DMB) son variantes alélicas de la misma enfermedad recesiva ligada al X, debidas a mutaciones en el gen de la distrofina, ubicado en el brazo corto de este cromosoma. La DMD afecta a 1 de cada 3,500 varones nacidos vivos, mientras que la variante menos severa (DMB) se presenta en 1 de cada 7,000 niños1. Aunque las funciones de esta proteína no están esclarecidas por completo, se sabe que participa en procesos de señalización intracelular, control del calcio intracelular y anclaje de la membrana del sarcolema, lo cual permite una buena y eficiente contracción muscular. Su ausencia produce deterioro muscular progresivo y la muerte hacia la segunda década de la vida en la DMD y hacia la quinta década en la DMB2, 3.
Esta enfermedad sin cura conocida, genera discapacidad en los afectados y un alto impacto económico y social en sus familias. Por ello se ha hecho un gran esfuerzo en el diagnóstico de portadoras para ofrecer alternativas reproductivas como la donación de óvulos, adopción, diagnóstico pre-implantación entre otras.
El diagnóstico de portadoras se ha hecho mediante diversos métodos tanto directos como indirectos; dentro de los métodos directos se tiene el southern blott, la determinación de dosis génica mediante densitometría, análisis de secuenciación y más recientemente la técnica de amplificación con sondas múltiples que dependen de ligación (MLPA)4-7. Dentro de los métodos indirectos se han utilizado los RFLPs (fragmentos de restricción de longitud polimórfica) y los STRs (short tandem repeats), cuya informatividad en ambos casos depende de su heterocigocidad, la cual varía para las diversas poblaciones; la mayor dificultad del análisis de ligamiento mediante los RFLPs son los casos de recombinación8. Dentro del gen de la distrofina, se han localizado un gran número de STRs que se ubican a lo largo del gen y en sus regiones extremas; esto contribuye a disminuir el riesgo de no descubrir casos de recombinación.
Hay estudios9, 10 que informan el uso de STRs con heterocigocidades que oscilan entre 37% y 93.3%9, 10, su uso se ha extendido particularmente en las familias donde no se demuestra deleción ni duplicación, Millar et al.11, lograron establecer haplotipos y el diagnóstico indirecto en 39% de las familias analizadas, con polimorfismos CA en la región 3’; Ferreiro et al.12, establecieron el estado de portadora o no portadora en 75% de las 52 familias analizadas al emplear 11 STRs.
El objetivo de este trabajo fue establecer mediante análisis de ligamiento y construcción de haplotipos, el estado de portadora o no portadora en 37 familias con enfermos de DMD y determinar si en algunas mujeres se observa la ausencia de un alelo que debería portar obligatoriamente, lo que se conoce como pérdida de heterocigocidad, y que indique deleción de este segmento del gen de la distrofina.
MÉTODOS
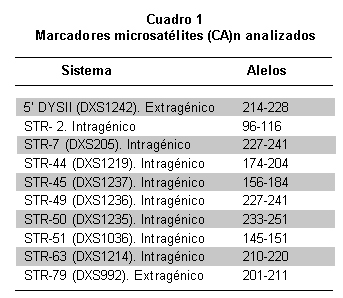
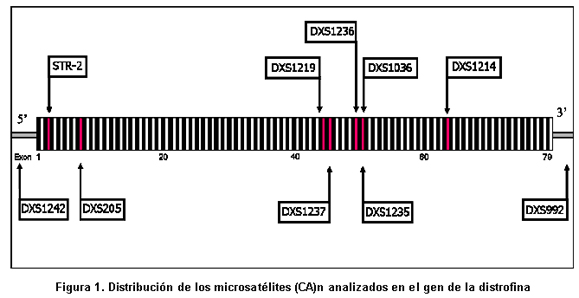
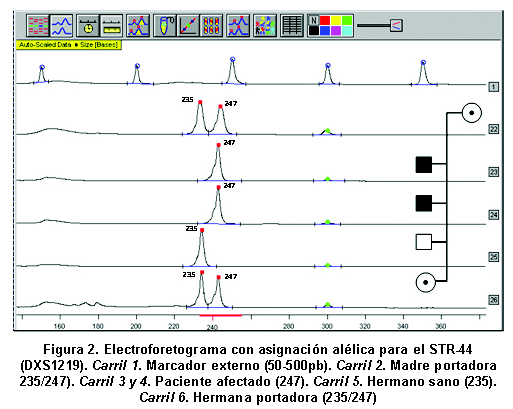
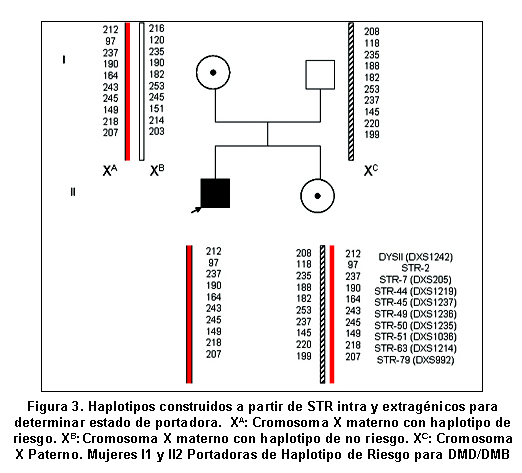
Para identificar las portadoras de DMD mediante análisis indirecto, se estudiaron 174 personas pertenecientes a 37 familias con pacientes afectados de DMD. En 19% de los casos hubo deleción en uno o varios exones del gen de la distrofina, según los informes de Silva et al.13 El 73% de los casos esporádicos, sin antecedentes familiares de esta entidad. En la población total se estudiaron 43 afectados, 102 mujeres familiares por línea materna y 29 hombres sanos. Previo consentimiento informado, aceptado por el Comité de Ética del Instituto de Ciencias Básicas de la Facultad de Medicina de la Universidad del Rosario, y según los lineamientos de la Resolución 8430 de 1993 del Ministerio de Protección Social, se tomaron muestras de sangre para extracción de ADN mediante el kit GFX (Invitrogen). En algunos casos se usaron muestras que reposan en el banco de ADN del laboratorio de Biología Molecular y Celular de la Universidad del Rosario, provenientes de personas que autorizaron su uso en la investigación. Se estandarizó la reacción en cadena de la polimerasa (PCR) para la amplificación de diez microsatélites o STRs dinucleotídicos con unidad de repetición (CA)n situados a lo largo del gen de la distrofina, dos fueron extragénicos y ocho intragénicos (Cuadro 1 y Figura 1). La PCR se hizo con los primers previamente descritos14. Para los STR DXS1242 y DXS992 se usó una mezcla de PCR a 20 μl, con reactivos de Promega para mantener las siguientes concentraciones: amortiguador 1X, MgCl2 3mM, DNTP 0.8mM, Taq DNA polimerasa 3U y 300ng de DNA. Para el resto de los microsatélites analizados se modificó la concentración de MgCl2 a 1.5mM, DNTP 0.2mM y 1.2 U de Taq DNA polimerasa. Las reacciones de PCR para 9 microsatélites se hicieron en el termociclador MJ research, a 94ºC por 6 minutos, 32 ciclos de 94ºC por 30 seg, 50ºC por 30 seg, 72ºC por 30 seg y 72ºC por 5 min. El STR-50 (DXS1235) se amplificó con 25 ciclos de: 94ºC por 30 seg, 62ºC por 30 seg, 65ºC por 2 min y 65ºC por 7 min. En cada montaje se usó un control negativo de amplificación donde se agregaba agua en vez de ADN con el objeto de verificar la presencia de posibles contaminantes de PCR. Los productos amplificados se sembraron sobre geles de poliacrilamida desnaturalizante (Reprogel® Pharmacia) y se corrieron en electroforesis vertical durante 200 minutos a 1500V, 60mA y 50°C en el secuenciador semiautomático ALF EXPRESS. El análisis se efectuó con las aplicaciones ALFWin y Allelinks, que permitieron asignar los alelos, según el tamaño en pares de bases de los productos obtenidos para cada individuo analizado. En cada corrido se usaron marcadores externos de 50-500pb (Amersham) e internos 250 ó 300pb (Amersham) que permitían asignar el alelo (Figura 2). La información alélica se organizó y se analizó para cada familia y se realizó la construcción del haplotipo para cada miembro del estudio, tomando como base el haplotipo de riesgo presente en el afectado (Figura 3).
Con base en los haplotipos, se determinó mediante recuento directo el número de mujeres que compartían el haplotipo de riesgo y que por tanto eran portadoras. Mediante asesoramiento genético personalizado se entregó a cada participante su resultado y en los casos de las mujeres portadoras de DMD se les informó acerca de los riesgos de recurrencia y sus opciones reproductivas.
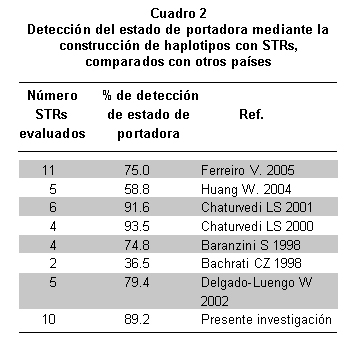
La heterocigocidad para cada sistema analizado se determinó mediante conteo directo en las mujeres de la filial 1. El porcentaje de portadoras identificadas con la técnica propuesta se comparó con el descrito en otras poblaciones (Cuadro 2).
RESULTADOS
Con los métodos descritos se determinó el estado de portadora en 89.2% de las mujeres estudiadas, de ellas 67 (65.7%) eran portadoras y 29 (23.5%) no portadoras. Las mujeres que poseen el haplotipo de riesgo que hay en los afectados se consideran como portadoras, y tienen una probabilidad de 50% de tener hijos varones con DMD.
El origen del haplotipo ligado a la enfermedad se pudo determinar en 11 familias, gracias a la posibilidad de analizar abuelas y/o abuelos maternos del enfermo. En 7 casos, el haplotipo lo transmitió la abuela y en 4 el abuelo.
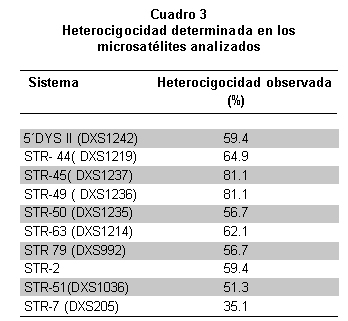
Los STRs utilizados mostraron heterocigocidad entre 35.1% y 81.1% (Cuadro 3). Los STR-45 y 49, fueron los más informativos y el STR-7 presentó la mayor homocigocidad en la población de estudio. En 10.8% de las familias fueron informativos sólo dos sistemas, en 8.1% lo fueron nueve, y en el resto entre 4 y 6 STRs, por lo que se puede considerar éste como el mínimo por usar en el momento de hacer este tipo de análisis.
DISCUSIÓN
Las DMD/DMB son entidades frecuentes, progresivas y letales, para las que no se ha logrado, hasta la fecha, un tratamiento efectivo. Por tener un modo de herencia ligado al X recesivo, las mujeres portadoras tendrán un riesgo de 50% de tener hijos varones afectados e hijas portadoras. La prevención primaria de DMD/DMB se basa en identificar e informar a las mujeres el riesgo de heredar la enfermedad a su descendencia.
Para establecer estado de portadora se pueden seguir distintos métodos a fin de evitar el error que se genera en el análisis molecular, cuando la causa de la DMD/DMB es la deleción o duplicación de uno o más exones, donde la presencia de una copia del gen normal a pesar de la presencia o ausencia de la mutante en las portadoras, oculta y hace que la mutación no sea fácilmente evidenciable. Por ejemplo, si un niño sufre DMD/DMB, muestra deleción del exón 19 y su madre es portadora, ella será heterocigota, pues un alelo presentará el exón 19 mientras que el otro no. Si se hace en la madre el análisis molecular, se tendrá banda de amplificación para el exón 19 que enmascara el diagnóstico del segmento ausente.
Por esta razón algunos autores15 utilizan la técnica de hibridación fluorescente in situ (FISH), con sondas específicas que señalan la presencia o ausencia de un exón sobre el alelo, método de costo económico considerable y útil sólo cuando el paciente presenta una deleción16; de hecho, según Silva et al.13 y en una elevada proporción de casos de otras latitudes, sólo 31% de los pacientes presentaron deleciones, por esto se considera el FISH como una alternativa poco informativa para la población colombiana de enfermos y sus familiares.
Otro método para reconocer portadoras consiste en el análisis de la dosis génica, con densitometría o mediante PCR en tiempo real14, el cual de nuevo se limitará apenas a los pacientes cuya mutación sea deleción o duplicación. Por otra parte, la secuenciación completa del gen de la distrofina sería una alternativa válida para identificar cualquier tipo de mutación en los enfermos, aunque no permite identificar a ninguna mujer portadora de deleciones o duplicaciones, además de ser una técnica costosa17.
Como se mencionó antes, un número considerable de casos de DMD/DMB no presentan deleción, por lo que se necesitan análisis de mutaciones de tipo indirecto, por ejemplo, la construcción de haplotipos mediante el estudio de regiones polimórficas al interior o alrededor del gen de interés, como en efecto son los STRs18-22.
Cada STR presenta varios alelos que permiten construir haplotip
os e identificar el haplotipo de riesgo para así reconocer a las mujeres portadoras del haplotipo que están en riesgo de transmitir la DMD/DMB a su descendencia (análisis indirecto de una mutación).
En el presente estudio, se logró determinar el estado de portadora en un alto número de mujeres analizadas (89.2%); esto permitió el asesoramiento acerca de los riesgos de tener hijos afectados o hijas portadoras e indicar estrategias reproductivas alternas, que constituyen la única forma de prevenir esta enfermedad altamente discapacitante y sin tratamiento conocido23, 24. Los análisis indirectos en otras poblaciones mundiales indican valores que oscilan entre 36.5% y 93.5%; con evaluación de entre 4 y 6 STRs (Cuadro 3)8, 12, 25-28; estos resultados se deben analizar con cautela, pues con el uso de pocos STRs y solamente intragénicos se puede perder la evidencia de procesos de recombinación, que son comunes a lo largo del gen de la distrofina por su gran longitud (2.4Mb), con valores de 9% a 12%28. Una recombinación entre el marcador utilizado y el exón de interés puede causar errores diagnósticos porque una mujer portadora con el haplotipo de riesgo podría no tener la mutación en el exón de interés o, por el contrario, puede poseer el haplotipo no ligado a la mutación y portar la enfermedad29, 30, esta situación obliga a establecer como indeterminada la condición o no de portadora en las mujeres con evidencia de recombinación.
La adecuada asignación de portadoras o no portadoras a través de haplotipos construidos con STRs se logra gracias a la informatividad de los sistemas utilizados, que depende de la heterocigocidad de cada uno de ellos; los sistemas que mostraron un alta informatividad en este estudio también se informaron con alta heterocigocidad en otras poblaciones25, 28, 29. Los sistemas como el STR-63 (DXS1214), STR-2 y STR-79 (DXS992), poco usados en el análisis de portadoras, demostraron una alta heterocigocidad, lo que determina que se deberían utilizar para la construcción de haplotipos. La informatividad de estos sistemas supera incluso la del STR-50 que se usa comúnmente para este fin. En Colombia una publicación previa31 hecha con sólo 2 STRs intragénicos demostró una baja eficiencia de 32% en identificar portadoras de DMD/DMB.
El origen del haplotipo de riesgo se logró determinar en algunas familias (10.8%) y fue predominantemente masculino (63.6%), resultado acorde con lo que informa la literatura donde se asocian los surgimientos de las mutaciones causantes de DMD/DMB a sucesos durante la espermatogénesis del abuelo materno del caso afectado. Como el error de replicación durante la división celular es la mayor fuente de mutación, el alto número de divisiones celulares durante la espermatogénesis lleva a que éstas se generen sobre todo en la línea germinal paterna. La continua producción de esperma durante la vida adulta puede implicar que las células germinales masculinas sean más sensibles a envejecimiento y senescencia por varios factores, por ejemplo acumulación de mutaciones en genes comprometidos en la maquinaria de reparación. Por último, la división celular en oogénesis es sólo prenatal, mientras que muchas divisiones en espermatogénesis ocurren luego de la madurez sexual. La mutación generada en la línea germinal paterna, se puede transmitir a sus hijas, y determina que ellas sean portadoras8, 32.
El análisis indirecto mediante construcción de haplotipos con 2 STRs extragénicos y 8 intragénicos permitió establecer el estado de portadora en una gran proporción de la población analizada de mujeres, que permite dar asesoramientos genéticos adecuados. Como en estudios previos hechos en la población colombiana de enfermos se determinó una baja proporción de deleciones, el análisis indirecto responde a la necesidad de identificar portadoras mediante la asignación del haplotipo de riesgo o ligado a la enfermedad, y permite ofrecer a estas familias una explicación de los riesgos y de las opciones reproductivas.
REFERENCIAS
1. Emery AE, Rimoin DL. Principles and practice of medical genetics. 2a ed. New York: Churchill Livingstone; 1990. [ Links ]
2. Roberts R. Dystrophins and dystrobrevins. Genome Biol. 2001; 2: 3001-6. [ Links ]
3. Acharyya S, Villalta A, Bakkar N, Bupha-Intr T, Janssen P, Carathers M, et al. Interplay of IKK/NF-kB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007; 117: 889-901. [ Links ]
4. Van Essen AJ, Kneppers AL, Van Der Hout AH, Scheffer H, Ginjaar IB, Ten Kate LP, et al. The clinical and molecular genetic approach to Duchenne and Becker muscular dystrophy: an updated protocol. J Med Genet. 1997; 34: 805-12. [ Links ]
5. Schwartz M, Dun M. Improved molecular diagnosis of dystrophin gene mutations using the multiplex ligation-dependent probe amplification method. Genet Test. 2004; 8: 361-7. [ Links ]
6. Freund AA, Scola RH, Arndt RC, Lorenzoni PJ, Kay CK, Werneck LC. Duchenne and Becker muscular dystrophy: a molecular and immunohistochemical approach. Arq Neuropsiquiatr. 2007; 65: 73-6. [ Links ]
7. Taylor PJ, Maroulis S, Mullan GL, Pedersen RL, Baumli A, Elakis G, et al. Measurement of the clinical utility of a combined mutation detection protocol in carriers of Duchenne and Becker muscular dystrophy. J Med Genet. 2007; 44: 368-72. [ Links ]
8. Delgado-Luengo WN, Borjas-Fuentes L, Zabala-Fernández W, Fernández-Salgado E, Solís-Añez E, Chávez C, et al. Detección de portadoras de distrofia muscular Duchenne/Becker a través del análisis de loci STRs ligados al gen de la distrofina en familias venezolanas. Invest Clin. 2002; 43: 239-54. [ Links ]
9. Oudet C, Heilig R, Mandel JL. An informative polymorphism detectable by polymerase chain reaction at the 3' end of the dystrophin gene. Hum Genet. 1990; 84: 283-5. [ Links ]
10. Clemens PR, Fenwick RG, Chamberlain JS, Gibbs RA, Andrade M de, Chakraborty R, et al. Carrier detection and prenatal diagnosis in Duchenne and Becker muscular dystrophy families, using dinucleotide repeat polymorphisms. Am J Hum Genet. 1991; 49: 951-60. [ Links ]
11. Miller M, Boehm C, Cotton M, Kazazian H. Usefulness of a CACA repeat polymorphism in genotype assignments in Duchenne/Becker muscular dystrophy. Am J Med Gen. 1992; 44: 473-6. [ Links ]
12. Ferreiro V, Szijan I, Giliberto F. Detection of germline mosaicism in two Duchenne muscular dystrophy families using polymorphic dinucleotide (CA)n repeat loci within the dystrophin gene. Mol Diagn. 2004; 8: 115-21. [ Links ]
13. Leiden Muscular Dystrophy pages© Center for Human and Clinical Genetics, Leiden University Medical Center. http:// www.dmd.nl [ Links ]
14. Silva CT, Fonseca D, Restrepo CM, Contreras N, Mateus H. Deleciones en el gen de la distrofina en 62 familias colombianas: correlación genotipo-fenotipo para la distrofia muscular de Duchenne y Becker. Colom Med. 2004; 35: 191-8. [ Links ]
15. Ligon AH, Kashork CD, Richards CS, Shaffter LG. Identification of female carriers for Duchenne and Becker muscular dystrophies using a FISH-based approach. Euro J Hum Genet. 2000; 8: 293-8. [ Links ]
16. Joncourt F, Neuhaud B, Jostarndt K, Kleinle S, Steiner B, Gallati S. Rapid identification of female carriers of DMD/BMD by quantitative real time PCR. Hum Mut. 2004; 23: 385-91. [ Links ]
17. Flanigan KM, Niederhausern A, Dunn DM, Alder J, Mendell JR, Weiss RB. Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet. 2003; 72: 931-9. [ Links ]
18. Percesepe A, Ferrari M, Coviello D, Zanussi M, Castagni M, Neri I, et al. Detection of a novel dystrophin gene mutation through carrier analysis performed during prenatal diagnosis in a case with intragenic recombination. Prenat Diagn. 2005; 25: 1011-14. [ Links ]
19. Alcántara MA, García-Cavazos R, Hernández-U E, González-Del Angel A, Carnevale A, Orozco L. Carrier detection and prenatal molecular diagnosis in a Duchenne muscular dystrophy family without any affected relative available. Ann Genet. 2001; 44: 149-53. [ Links ]
20. Ferreiro V, Giliberto F, Frangipane L, Szijan I. The role of polymorphic short tandem (CA)n repeat loci segregation analysis in the detection of Duchenne muscular dystrophy carriers and prenatal diagnosis. Mol Diag. 2005; 9: 67-80. [ Links ]
21. Bachrati CZ, Somodi Z, Endreffy E, Kalmar T, Rasko I. Carrier Detection by microsatellite analysis of Duchenne/Becker muscular dystrophy in Hungarian families. Ann Hum Gent. 1998; 62: 511-20. [ Links ]
22. Montejo Y, Zaldívar VT, Acevedo A, Guerra N. Diagnóstico prenatal y de portadores en familia con distrofia muscular de Duchenne. Introducción de nuevos marcadores. Rev Neurol 2001; 33: 1094-95. [ Links ]
23. Chakkalakal, JV, Thompson J, Parks RJ, Jasmin B. Molecular, cellular, and pharmacological therapies for Duchenne/Becker muscular dystrophies. FASEB. J 2005; 19: 880-91. [ Links ]
24. Fall AM, Johnsen R, Honeyman K, Iversen P, Fletcher S, Wilton SD. Induction of revertant fibres in the mdx mouse using antisense oligonucleotides. Genet Vaccines Ther. 2006; 4: 3-15. [ Links ]
25. Chaturvedi LS, Srivastava S, Mukherjee M, Mittal B. Analysis of dinucleotide repeat loci of dystrophin gene. Indian J Med Res. 2001; 113: 19-25. [ Links ]
26. Chaturvedi LS, Mittal RD, Srivastava S, Mukherjee M, Mittal B. Analysis of dinucleotide repeat loci of dystrophin gene for carrier detection, germline mosaicism and de novo mutations in Duchenne/Becker muscular dystrophy. Clin Genet. 2000; 58: 234-5. [ Links ]
27. Baranzini SE, Giliberto F, Dalamon V, Barreiro C, García-Erro M, Grippo J, et al. Carrier detection in Duchenne and Becker muscular dystrophy Argentine families. Clin Genet 1998; 54: 503-11. [ Links ]
28. Bachrati CZ, Somodi Z, Endreffy E, Kalmár T, Raskó I. Carrier detection by microsatellite analysis of Duchenne/Becker muscular dystrophy in Hungarian families. Ann Hum Genet. 1998; 62: 511-20. [ Links ]
29. Carsana A, Frisso G, Tremolaterra MR, Ricci E, De Rasmo D, Salvatore F. A larger spectrum of intragenic short tandem repeats improves linkage analysis and localization of intragenic recombination detection in the dystrophin gene: an analysis of 93 families from southern Italy. J Mol Diagn. 2007; 9: 64-9. [ Links ]
30. Chavarría G, Reis A, Azofeifa J. Determinación indirecta mediante marcadores de ADN del estado de portadora de distrofia muscular de Duchenne (DMD) en una familia costarricense. Acta Pediat Costarric. 2002; 16: 1-12. [ Links ]
31. Hernández P, Gómez Y, Restrepo C. Identificación de portadoras de distrofia muscular de Duchenne y Becker (DMD/DMB) mediante análisis de dosis génica y polimorfísmos de ADN. Biomedica. 2000; 20: 228-37. [ Links ]
32. Ellegren H. Characteristics, causes and evolutionary consequences of male-biased mutation. Proc R Soc B. 2007; 274: 1-10. [ Links ]

