










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkColombia Médica
versão On-line ISSN 1657-9534
Colomb. Med. v.39 supl.2 Cali jun. 2008
Deficiencia de glucosa 6 fosfato deshidrogenasa: análisis enzimático y molecular en una población de Bogotá
Magda Carolina Sánchez, MSc1, Victoria Eugenia Villegas, MSc1, Dora Fonseca, MSc2
1. Profesora Asistente, Unidad de Morfología, Facultad de Medicina, Universidad del Rosario, Bogotá, Colombia. e-mail: msanche@urosario.edu.co victoria.villegasga@urosario.edu.co
2. Profesora Principal, Unidad de Genética, Facultad de Medicina, Universidad del Rosario, Bogotá, Colombia. e-mail: dfonseca@urosariuo.edu.co
Recibido para publicación junio 25, 2007 Aceptado para publicación abril 18, 2008
RESUMEN
Objetivo: Determinar qué tan frecuente es la deficiencia de glucosa 6 fosfato deshidrogenasa (G6PD) y realizar análisis molecular para identificar las variantes A+, A- y mediterránea en una población de residentes en Bogotá.
Métodos: Se analizaron 348 personas que residen en Bogotá, pertenecientes a la Policía Nacional y a la Universidad del Rosario. La actividad enzimática se determinó en muestras de sangre mediante espectrofotometría con el kit Trinity Biotech (Cat 345-B). Los valores de hemoglobina (Hg) y hematócrito (Hto) se determinaron con el método de Drabkin y sedimentación, respectivamente. La determinación de las variantes moleculares se realizó mediante amplificación por reacción en cadena de la polimerasa (PCR) y análisis de fragmentos de restricción de longitud polimórfica (RFLP) con las enzimas NlaIII, Fok I y MboII para A+, A- y mediterránea, respectivamente. Se hicieron análisis estadísticos para comparar la concentración de Hg en personas sanas y deficientes, la actividad de G6PD por géneros y los datos de frecuencia a nivel mundial.
Resultados y conclusiones: La frecuencia de deficiencia de G6PD para la población en estudio fue 3.1%. En 1.4% de los casos se observó actividad deficiente, en 1.7% actividad intermedia y en 0.6% actividad aumentada. No se encontraron las variantes moleculares A+, A- y mediterránea en ningún afectado. La actividad de G6PD no tuvo diferencias por género. Se encontró diferencia significativa en el valor de hemoglobina entre las personas sanas y deficientes de G6PD. Los individuos deficientes eran asintomáticos lo que indica mecanismos de compensación de estrés oxidativo. Las mujeres deficientes son heterocigotos con una inactivación preferencial del cromosoma X anormal y al ser portadoras tienen riesgo de 50% de tener hijos afectados con la enfermedad. La identificación de mujeres y hombres deficientes permite establecer medidas preventivas ante posibles crisis hemolíticas desencadenadas por infecciones y drogas.
Palabras clave: Eritrocito; Genética; NADPH; Deficiencia; Anemia hemolítica.
Glucose-6-phosphate dehydrogenase deficiency: enzimatic and molecular analysis in a Bogotá population
SUMMARY
Objective: To determine the frequency of G-6PD and molecular analysis for identification of A+, A- and Mediterranean in healthy persons in Bogotá.
Methods: Quantitative spectrophotometric assays for enzyme activity of G-6PD were carried out on the red cells of 348 asymptomatic and healthy adult males and females. Through molecular analysis of DNA from G-6PD deficients the relevant exons were amplified for PCR and then analysed with the restriction enzymes NlaIII, Fok I and MboII, for the detection of A+, A- and Mediterranean variants.
Results and conclusions: Among 348 samples, 1.4% exhibited total deficiency and 1.7% had intermediate deficiency while 96.3% were normal. The combined prevalence was 3.7%. In enzymatic activity no statistically significance was seen between males and females. No variant was found among these patients and any of the subjects studied displayed any sign of hemolysis and other clinical manifestations. Although it is not yet clearly understood other mechanisms must exist to offer protection from the oxidative stresses. The finding of severe enzyme deficiency in some heterozygote females is due to extreme degree of X inactivation of the normal chromosome.
Keywords: Erythrocyte; Genetics; NADPH; Glucose-6-phosphate dehydrogenase deficiency; Hemolytic anemia.
La deficiencia de glucosa 6 fosfato deshidrogenasa (G6PD; EC 1.1.1.49) es el defecto enzimático congénito más frecuente y afecta alrededor de 400 millones de personas en el mundo1. La enzima cataliza el paso de entrada de glucosa-6-fosfato en la vía de las pentosas, produce su oxidación a 6-fosfogluconolactona y proporciona al glóbulo rojo nicotinamida adenina dinucleótido fosfato reducido (NADPH), que se requiere para la acción normal de la metahemoglobina reductasa y el mantenimiento de un nivel adecuado de glutatión reducido (GSH), forma en la que puede proteger a los eritrocitos del daño oxidativo y disminuir la susceptibilidad a hemólisis2, 3. Aunque casi todos los individuos con la deficiencia enzimática son asintomáticos, este defecto puede causar ictericia neonatal, y anemia hemolítica desde leve hasta crónica, no esferocítica, con episodios de crisis severas, inducidas por infecciones, drogas específicas o consumo de habas4. El gen de la G6PD se localiza en la región terminal del brazo largo del cromosoma X (Xq28), cerca a los genes de la hemofilia A y la disqueratosis congénita. La deficiencia se transmite como un rasgo recesivo ligado a X, por lo que los hombres son hemicigotos, normales o afectados, y las mujeres son homocigotas normales o deficientes y heterocigotas.
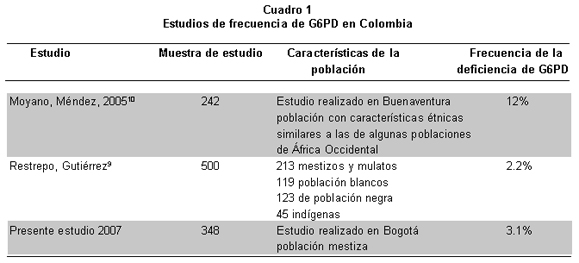
La diferenciación genotípica a partir de la expresión fenotípica es difícil de hacer, por el fenómeno de inactivación del cromosoma X5, 6. En el mundo se han caracterizado cerca de 400 variantes bioquímicas de la G6PD, las más frecuentes son la B (actividad normal), A+ (actividad normal), A- (8%-20% de actividad normal), y mediterránea (<5% de la actividad normal)7. La deficiencia en los eritrocitos se encuentra por lo general en áreas donde la malaria es o ha sido endémica, y su frecuencia se estudió ampliamente en varios países de donde se han informado valores que oscilan entre 0.39% y 65%, con variaciones determinadas por el origen étnico. En Latinoamérica la población más estudiada ha sido la del Brasil8 con frecuencias hasta de 8%. En Colombia Restrepo y Gutiérrez9 en 1968 estudiaron por primera vez la deficiencia de G6PD y analizaron 500 individuos sanos clasificados como población mestiza, blanca, negra e indígena. Según estos autores la deficiencia de G6PD afectaba 2% de la población mestiza y cerca de 22% de la población negra. Moyano y Méndez6, informaron una prevalencia alta de la deficiencia (12%) en una muestra de 242 personas con malaria que acudían al Programa de Enfermedades Tropicales en la ciudad de Buenaventura (Cuadro 1). La alta frecuencia hallada se explica por el efecto protector de la deficiencia de G6PD en personas con infección por malaria8-10. La presente investigación determinó qué tan frecuente es la deficiencia de G6PD en una población de hombres y mujeres de Bogotá, mediante evaluación por espectrofotometría de la actividad enzimática. Las personas con actividad deficiente o intermedia se estudiaron con la reacción en cadena de la polimerasa (PCR) y el análisis de fragmentos de longitud polimórfica (RFLP), para identificar las variantes A+, A- y mediterránea, que se deben a mutaciones puntuales del gen de la G6PD. La variante A+ se produce por el cambio de adenina por guanina en la posición 376 (376 A-G) del exón 4, en la A- se dan dos transiciones: 376 A-G y 202 G-A en los exones 4 y 5 y en la mediterránea el cambio se genera en la posición 563 del exón 6 y corresponde a una transición de citosina por timina (563 C-T). La determinación de la frecuencia de la deficiencia permite analizar su impacto en salud pública y descubrir individuos en riesgo de sufrir crisis hemolíticas ante desencadenantes de la enfermedad o mujeres en riesgo de tener hijos afectados, para así dar asesoramiento genético y pautas de manejo frente a complicaciones severas desencadenadas por factores externos o en el manejo de la ictericia neonatal grave que se presenta en los recién nacidos con deficiencia.
MÉTODOS
Para el análisis enzimático y molecular de la deficiencia de G6PD, se estudiaron 348 residentes en Bogotá, 174 hombres y 174 mujeres. Previo consentimiento informado, aceptado por el Comité de Ética de la Facultad de Medicina de la Universidad del Rosario y de acuerdo con lo dispuesto en la Resolución 8430 de 1993 del Ministerio de Salud, se tomaron 10 ml de sangre periférica anticoagulada con EDTA de cada participante. Las muestras correspondían a voluntarios de la Policía Metropolitana de Bogotá y a estudiantes de la Universidad del Rosario, quienes se incluyeron por su participación voluntaria en el estudio y evidencia de no tener manifestaciones clínicas de desórdenes hemolíticos.
Análisis enzimático de la actividad de la G6PD. La actividad enzimática de la G6PD se determinó mediante el kit Trinity Biotech (Cat 345-B). Este kit permite una determinación cuantitativa y cinética en sangre a 340 nm. En cada medición se utilizaron controles con actividad deficiente, intermedia y baja, suministrados por la casa comercial. El protocolo para esta determinación se desarrolló de la manera sugerida en el kit. Las lecturas finales de absorbancia se hicieron en el espectrofotómetro TECAN.
La actividad de la G6PD se expresó en forma U/g de hemoglobina (Hb), por lo que se efectuó también la determinación de Hb. Con este fin, se utilizó el método colorimétrico, que se fundamenta en la formación de cianuro de hemoglobina, cuando esta molécula responde al reactivo de Drabkin. Como control de la hemoglobinometría se utilizó el Standard Hemoglo-Wiener. La lectura se hizo en un espectrofotómetro 21 D de Milton Roy, con mediciones a 540 nm.
Análisis molecular para demostrar las variantes A+, A- y mediterránea. A los individuos con actividad deficiente e intermedia de G6PD, se les realizó el análisis molecular de las variables A+, A- y mediterránea, para lo cual fue necesario extraer ADN a partir de sangre periférica mediante el protocolo estándar7. El ADN obtenido se verificó en geles de agarosa al 2%, teñidos con bromuro de etidio y visualizados sobre transiluminador.
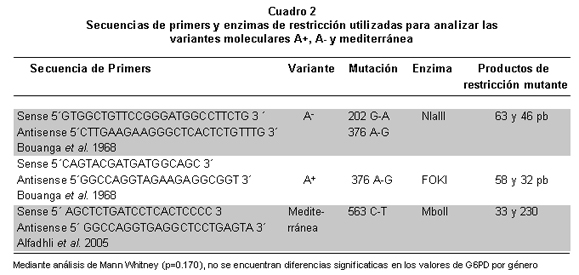
Para amplificar las regiones que permiten describir la presencia de las variantes A+ y A-, se utilizaron los iniciadores descritos por Bouanga et al.11 (Cuadro 2). La amplificación se realizó en un termociclador PTC 100 MJ Research con el siguiente programa: 95ºC por 5 min 35 ciclos a 95ºC por 20 seg, 62 ºC por 20 seg y 72ºC por 20 seg, además de una extensión final a 72ºC por cinco minutos. Para la identificación de la variante mediterránea se utilizaron los primers descritos por Alfadhli et al.12 (Cuadro 2). La mezcla y condiciones de amplificación fueron similares a la anteriores excepto en que la concentración de MgCl2 varió a 2 mM y la temperatura de annealing se usó a 60ºC durante 45 segundos, tiempo que también se utilizó para la extensión parcial. En todos los montajes se empleó un control negativo de amplificación para monitorear la ausencia de contaminantes de PCR.
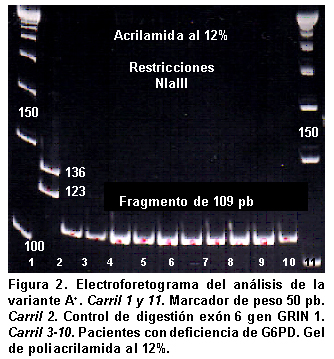
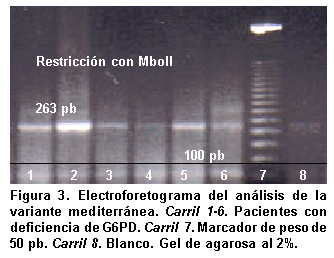
Los productos amplificados se verificaron en geles de agarosa al 2% y se sometieron a corte con las enzimas de restricción Nla III, Fok I y Mbo II (Biolabs) para la identificación de las variantes A+, A- y mediterránea, respectivamente (Cuadro 2). Las mezclas de digestión se realizaron a volumen final de 20 µl, con amortiguador 1X, 10 U de cada enzima y 10 µl de producto amplificado. La restricción se efectuó a 37°C durante 8 horas y la reacción se detuvo al agregar 5 µl de amortiguador de carga. Los productos digeridos se analizaron con electroforesis en geles de poliacrilamida al 12% y agarosa al 2%, y se tiñeron con bromuro de etidio, para describir la presencia o no de la variante de acuerdo con los tamaños en pares de bases observados y esperados de los productos de digestion (Cuadro 2).
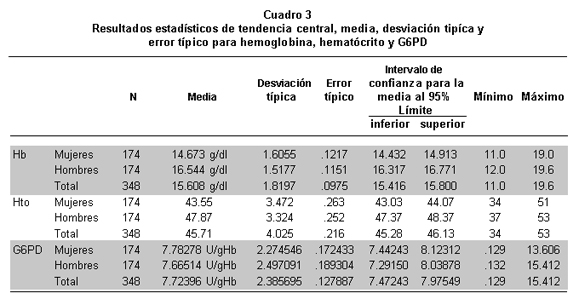
Análisis estadístico. Para las variables cualitativas de género y de clasificación de G6PD se utilizaron distribuciones de frecuencia y porcentuales y en las variables cuantitativas Hg, hematócrito y G6PD se usaron medidas de tendencia central: promedio y mediana y de dispersión: rango y desviación estándar (Cuadro 3).
Para evaluar si hay diferencias entre los promedios de Hb, Hto y G6PD con género se utilizó la prueba de comparación de medias, prueba t de Student, si cumplía con los supuestos de homogeneidad de variables (prueba de Levene) y normalidad (prueba de Kolmogorov Smirnorv y Shapiro Wilks), en caso de no cumplirse estos supuestos se utilizó la prueba no paramétrica de Mann Whitney para diferencia de medianas. Estas pruebas también se hicieron a fin de comparar los grupos de población normal para la actividad de la G6PD con el grupo de actividad disminuida o aumentada en relación con la Hb.
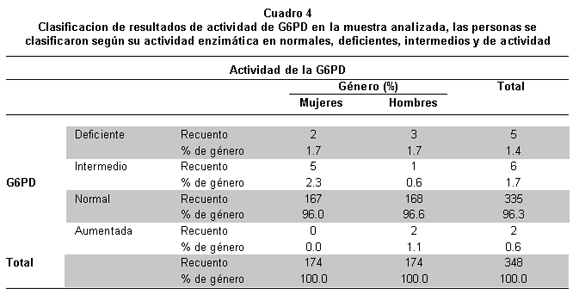
Para evaluar la clasificación de G6PD por género, se utilizó la prueba de diferencia de proporciones x2 (valores esperados >5) o prueba exacta de Fisher (valores esperados <5). Las pruebas estadísticas se evaluaron a un nivel de significancia del 5% (p<0.05) (Cuadro 4).
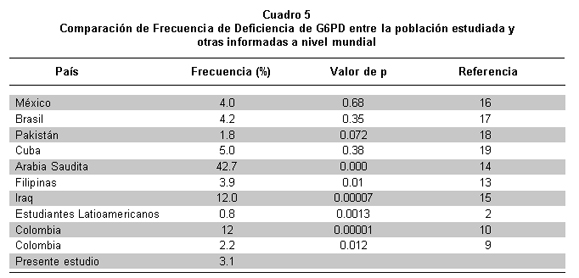
La comparación de la frecuencia de deficiencia de G6PD hallada en el presente estudio con la de otras poblaciones a nivel mundial se realizó mediante la tabla de contingencia de 2x2 con un intervalo de confianza del 95% p<0.05 (Cuadro 5).
RESULTADOS
En la población en estudio se encontró una frecuencia de deficiencia de G6PD de 3.1%, pues 11 de las 348 personas analizadas presentaron valores inferiores a los indicados como normales en el kit Trinity Biotech (4.6 a 13.5 U/g Hb).
Al tener como parámetro el valor enzimático del control deficiente provisto por el kit de cuantificación (hasta 2.1 U/gHb), 5 individuos (3 hombres y 2 mujeres) se clasificaron en este rango de deficiencia, lo que correponde al 1.4% de la poblacion analizada. La defi-ciencia se categorizó como intermedia en 6 personas (5 mujeres y 1 hombre), con valores enzimáticos entre 2.1 y 4.5U/g Hb, con una frecuencia de 1.7% (Cuadro 6).
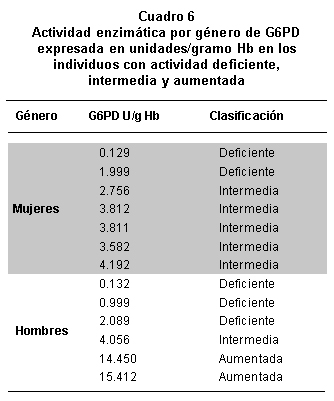
En 96.3% de los casos la actividad enzimática fue normal, mientras que 2 de los hombres estudiados (0.6%) presentaron valores enzimáticos que indican actividad aumentada.
Se encontraron diferencias significativas entre las medianas de Hb, siendo mayor en los hombres (16.5g/dl) que en las mujeres (14.6g/dl) (p<0.001, Mann Whitney): lo que muestra una tendencia a que la media de Hb y Hto sea mayor en el género masculino, (p<0.001), en cuanto a la actividad de la G6PD no hay diferencias entre género (p=0.170, Mann Whitney).
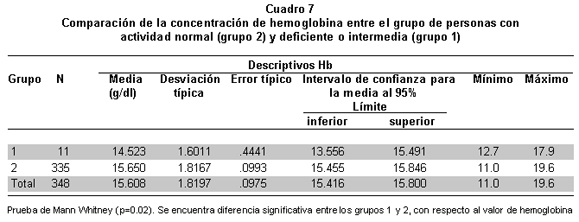
Se evidenció diferencia significativa entre las medianas de Hb de los dos grupos que fue mayor en los de actividad normal de G6PD (15.6) que en el grupo de actividad disminuida y aumentada (14.5) (p=0.020 Mann Whitney) (Cuadro 7).
Para la población en estudio se determinó una actividad normal expresada en U/g Hb de 7.78+2.2 en mujeres y 7.6+2.5 en hombres con un valor para la población general de 7.7+2.4.
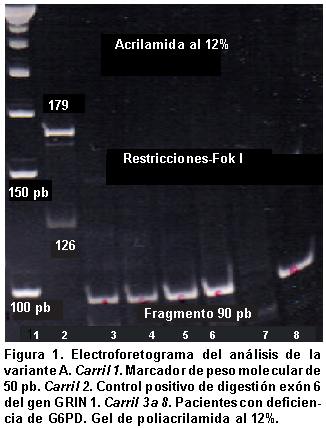
El análisis molecular mediante PCR y análisis de restricción de longitud polimorfica se realizó en las 11 personas con actividad enzimática menor a la normal y los análisis con electroforesis indicaron que ninguna de ellas portaba las variantes A+, A- y mediterránea, como se evidenció por la presencia de productos de 109, 90 y 263 pb respectivamente, que corresponden a alelos que no presentan estas mutaciones y que por tanto no generan sitios de reconocimiento para las enzimas de restricción NlaIII, Fok I y Mbo II (Figuras 1, 2, 3 y 4).

La comparación de la frecuencia de deficiencia en el presente estudio indicó diferencias estadísticamente significativas (p<0.05) con las poblaciones de Filipinas13, Arabia Saudita14, estudiantes latinoamericanos2, Irak15 y Buenaventura (Colombia)10, mientras que no se evidenciaron tales diferencias con México16, Brasil17, Pakistán18, Cuba19 y la población mestiza de Colombia9.
DISCUSIÓN
La deficiencia de G6PD constituye la enfermedad genética hereditaria que más afecta personas alrededor del mundo, y su frecuencia se ha analizado ampliamente en diferentes poblaciones.
En el presente estudio se determinó que 1.7% de las personas participantes presentaban actividad deficiente y 1.4% tenían actividad intermedia o parcial, es decir, una frecuencia combinada de 3.1%, similar a la informada en otros países como Cuba y México, pero con diferencias significativas en poblaciones como Arabia Saudita o Iraq, que han indicado frecuencias muy altas. Las diferencias poblacionales son el reflejo del impacto de la constitución genética en los distintos países, de tal manera que las mayores frecuencias se esperan en individuos con ascendencia africana y mediterránea, zonas endémicas para malaria o con altos grados de consanguinidad. En las Américas, la deficiencia es prevalente como resultado de migraciones en tiempos recientes5, 18. El único estudio que comunicó frecuencias de esta deficiencia enzimática en Colombia lo hicieron en 1968 Restrepo y Gutiérrez9 quienes indicaron para la población mestiza un valor de 2.2%, que no presenta diferencia estadística significativa (p<0.05) con los datos del presente estudio, en residentes de Bogotá, que tienen una mezcla étnica por las continuas migraciones que hacia la capital efectúan personas de diferentes acervos genéticos. Se ha establecido una asociación entre la prevalencia de defectos eritrocíticos y pacientes con diagnóstico de malaria, razón que explica la alta frecuencia que se informa para otras poblaciones colombianas como la de Buenaventura, donde se presenta la deficiencia de G6PD en 12% de los casos analizados10. La deficiencia confiere protección contra la malaria, y produce un efecto selectivo en poblaciones deficientes, esta presión selectiva se debe a la reducción en la concentración de NAPDH, secundaria a la deficiencia de G6PD, que es indispensable para la proliferación del parásito dentro de los eritrocitos. Hay estudios en África que indican reducción en el riesgo para contraer malaria severa hasta en 58%, en mujeres heterocigotas y hombres hemicigotos, observación debatida por otros autores que evidencian una mínima protección en mujeres heterocigotas18, 20. La deficiencia protege también de entidades como oclusión retinal (RVO), lo que produce efectos sobre la distribución de la enfermedad en el mundo21.
Se ha estimado que cerca de 7.5% de la población mundial porta el gen para la deficiencia de G6PD, y sin embargo la gran mayoría son clínicamente asintomáticos a lo largo de la vida, lo que determina que en poblaciones como la estudiada en Bogotá, que corresponde a personas sanas, se evidencien hallazgos de actividad deficiente, esta observación ha sido descrita por otros autores12, 14, 19, 22, lo que establece la existencia de mecanismos de compensación genéticos o no genéticos que ofrecen protección al estrés oxidativo, que tiene un papel importante en la expresión clínica de la enfermedad; de la misma manera permite discutir si la actividad enzimática puede ser un parámetro predictivo pobre de crisis hemolíticas agudas y no se correlaciona necesariamente con las características clínicas22,23. El hallazgo de mujeres con actividad deficiente o intermedia que para la población en estudio fue 63.6% del total de deficientes es una observación llamativa, pues al ser una entidad recesiva ligada al cromosoma X, se espera que los afectados sean los hombres quienes al ser hemicigotos sólo poseen una copia del gen. En las mujeres puede ocurrir selección celular somática a través del fenómeno de inactivación del cromosoma X, que es un evento en los mamíferos como mecanismo de compensación de dosis, ante la presencia de un solo cromosoma X en los hombres. En las mujeres de actividad deficiente, existe un mosaicismo en la población de glóbulos rojos con niveles normales y anormales de enzima, de tal forma que la aparición de heterocigotas con deficiencia profunda se debe a un grado extremo de inactivación del cromosoma X normal. Se ha considerado que las mujeres portadoras que no desarrollan manifestaciones clínicas deben tener mínimo 70% de glóbulos rojos normales5, 6,24.
En el presente estudio los pacientes con actividad deficiente y actividad intermedia evidenciaron diferencias significativas con el valor de Hg de los normales, lo que indica que análisis más detallados de índices biométricos podrían indicar alguna manifestación de este déficit enzimático. El hallazgo de mujeres homocigotas, con los dos alelos del gen de la G6PD alterados, un evento muy poco probable que se debe a mecanismos como disomia uniparental o ser hija de una mujer portadora y un hombre afectado. A pesar de la discusión planteada, la identificación de individuos afectados es importante porque permite ejercer medidas preventivas ante el posible desencadenamiento de manifestaciones graves como anemia hemolítica aguda en respuesta a estímulos como ingesta de habas, infecciones respiratorias virales, hepatitis, neumonía bacteriana e ingesta de diversos medicamentos como antimaláricos, sulfonamidas, nitrofuranos y antipiréticos; esta observación cobra mayor relevancia si se tiene en cuenta que parte de la población de este estudio correspondió a miembros de la policía que por su actividad laboral son susceptibles a ser tratados con antimaláricos, lo que puede exacerbar complicaciones hemolíticas5.
Las mujeres afectadas que son heterocigotas sufren el mismo efecto clínico que los hombres hemicigotos, y el conocimiento de su condición de portadora permite asesorar sobre el riesgo de tener hijos varones afectados con la enfermedad que es de 50% en cada gestación, éstos pueden ser asintomáticos o por el contrario sufrir la complicación neonatal más importante que es ictericia, que se asocia con daño neurológico irreversible; se ha establecido que la deficiencia de G6PD está presente hasta en 3% de los neonatos ictéricos4, 25. En los casos más severos la deficiencia es letal para los embriones26.
Dos de los hombres en el presente estudio se clasificaron con actividad enzimática aumentada, pues su evaluación de G6PD indicó un valor superior al rango normal establecido de 13.5 U/gHb, este hallazgo puede obedecer a la presencia de variantes que generen alteraciones en la velocidad de expresión de la enzima o su estabilidad frente a la degradación proteolítica. Además, se puede considerar una actividad aumentada en individuos cuyo sitio de procedencia difiera de la altura sobre el nivel del mar de Bogotá (2,600 m), porque en esta situación se produce estimulación de la corteza adrenal por la ACTH, que origina la secreción de cortisol. La liberación del cortisol es uno de los activadores más importantes de la gluconeogénesis, e interviene de manera directa en muchas reacciones de las vías glucolítica y de las pentosas27, principalmente la primera reacción catalizada por la G6PD, como respuesta integrada al estímulo hormonal externo, con aumento de la glucólisis que provoca a su vez un incremento en los sistemas enzimáticos que producen NADPH, por ejemplo G6PD.
En la población deficiente estudiada no se encontró ninguna de las variantes comunes como la A+, A- y mediterránea, por lo que se necesitan estudios complementarios como secuenciación que logren identificar si la alteración ocurre en otras regiones diferentes a las estudiadas. En diversas poblaciones del mundo se ha establecido una alta heterogeneidad molecular con identificación de hasta 27 genotipos diferentes en una misma población de afectados e incluso se ha sabido la presencia de variantes nuevas y privadas de un país determinado7, 9,13,16,19,28-30.
Los datos del presente trabajo corresponden a población mestiza residente en Bogotá, y se analizó un número superior de individuos de estas mismas características respecto al trabajo de Restrepo y Gutiérrez9 en 1968. La frecuencia de 3.1% indica que la deficiencia de G6PD puede ser muy notoria en el país y amerita estudios adicionales en una muestra representativa para Colombia que abarque todas las regiones, las cuales por sus diversos componentes étnicos tienen características genéticas diferentes, de la misma manera dada la heterogeneidad genotípica y fenotípica de la entidad es necesario ampliar el conocimiento para ofrecer adecuados asesoramientos genéticos a las portadoras y afectados, los cuales pueden pasar inadvertidos, pues son asintomáticos.
A pesar de no tener manifestaciones clínicas aparentes las personas con actividad enzimática deficiente o intermedia deficiente, son altamente susceptibles de desarrollar crisis hemolíticas desencadenadas por exposición a agentes oxidantes, lo cual sólo se puede prevenir mediante la identificación de estos individuos. La frecuencia de deficiencia de G6PD que se comunica en este trabajo alerta a las autoridades médicas en la instauración de filtrados selectivos que identifiquen los sujetos con esta deficiencia enzimática en poblaciones de riesgo, como los recién nacidos con ictericia no debida a incompatibilidad de grupo sanguíneo o población con acervo genético africano. En Colombia no ha habido ningún estudio molecular distinto al de la presente investigación, y el hecho de no encontrar las variantes moleculares más comunes A+, A-, y mediterránea, puede indicar la presencia de re-arreglos en el ADN nuevos y característicos de esta población, lo que se debe estudiar con análisis de secuenciación.
REFERENCIAS
1. Bonilla JF, Sánchez MC, Chuaire L. Glucosa 6 fosfato deshidrogenasa. Respuesta del eritrocito humano y otras células a la disminución en su actividad. Colom Med. 2007; 38: 76-83. [ Links ]Full text
2. Acosta T, Suárez M, Nuñez V, Marín L, Cordero A. Efecto hemolítico de la cloroquina en estudiantes deficientes de Glucosa-6-Fosfato-Deshidrogenasa. Rev Cubana Invest Biomed. 2003; 22: 180-5. [ Links ]
3. Fonseca D, Mateus H, Silva C, Contreras N, Restrepo C. Deficiencia de Glucosa 6-Fosfato Deshidrogenasa. Aspectos generales de la eritroenzimopatía más frecuente en el mundo. Acta Med Colomb. 2005; 30: 59-64. [ Links ]
4. Koosha A, Rafizadeh B. Evaluation of neonatal indirect hyperbilirubinaemia at Zanjan Province of Iran in 2001-2003: prevalence of glucose-6-phosphate dehydrogenase deficiency. Singapore Med. J 2007; 48: 424-8. [ Links ]
5. Luzzatto L. Glucose 6-phosphate dehydrogenase deficiency: from genotype to phenotype. Haematologica. 2006; 91: 1303-6. [ Links ]
6. Au WY, Lam V, Pang A, Lee WM, Chan JL, Song YQ, et al. Glucose-6-phosphate dehydrogenase deficiency in female octogenarians, nanogenarians, and centenarians. J Gerontol A Biol Sci Med Sci. 2006; 61: 1086-9. [ Links ]
7. Yan T, Cai R, Mo O, Zhu D, Ouyang H, Huang L, et al. Incidence and complete molecular characterization of glucose-6-phosphate dehydrogenase deficiency in the Guangxi Zhuang autonomous region of southern China: description of four novel mutations. Haematologica. 2006; 91: 1321-8. [ Links ]
8. Castro S, Weber R, Dadalt V, Tavares V, Giugliani R. Prevalence of G6PD deficiency in newborns in the south of Brazil. J Med Screen. 2006; 13: 85-6. [ Links ]
9. Restrepo AM, Gutiérrez E. The frequency of glucose-6-phosphate dehydrogenase deficiency in Colombia. Am J Hum Genet. 1968; 20: 82-5. [ Links ]
10. Moyano M, Méndez F. Defectos eritrociticos y densidad de la parasitemia en pacientes con malaria por Plasmodium falciparum en Buenaventura, Colombia. Rev Panam Salud Publica. 2005; 18: 25-31. [ Links ]
11. Bouanga JC, Mouele R, Prehu C, Wajcman H, Feingold J, Galacteros F. Glucose-6-phosphate dehydrogenase deficiency and homozygous sickle cell disease in Congo. Hum Hered. 1998; 48: 192-7. [ Links ]
12. Alfadhli S, Kaaba S, Elshafey A, Salim M, AlAwadi A, Bastaki L. Molecular characterization of glucose-6-phosphate dehydrogenase gene defect in the Kuwaiti population. Arch Pathol Lab Med. 2005; 129: 1144-7. [ Links ]
13. Padilla C, Nishiyama K, Shirakawa T, Matsuo M. Screening for glucose-6-phosphate dehydrogenase deficieny using a modified formazan method: a pilot study on Filipino male newborns. Pediatr Int. 2003; 45: 10-5. [ Links ]
14. AK Al-Ali. Common G6PD Variant from Saudi population and its prevalence. Ann Saudi Med. 1996; 16: 654-6. [ Links ]
15. Hassan MK, Taha JY, Al-Naama LM, Widad NM, Jasim SN. Frequency of haemoglobinopathies and glucose-6-phosphate dehydrogenase deficiency in Basra. East Mediterr Health J 2003; 9: 45-54. [ Links ]
16. Medina MD, Vaca G, López-Guido B, Westwood B, Beutler E. Molecular genetics of Glucose-6-Phosphate Dehydrogenase deficiency in Mexico. Blood Cells Mol Dis. 1997; 23: 88-94. [ Links ]
17. Compri MBm Saad ST, Ramalho AS. Genetico-epidemiological and molecular investigation of G-5-PD deficiency in a Brazilian community. Cad Saude Publica. 2000; 16: 335-42- [ Links ]
18. Mansoor N, Nadir A, Masood A, Mohammad A, Bhatti FA, Asif N. Frequency of glucose-6-phosphate dehydrogenase deficiency in some ethnic groups of Pakistan. JCPSP. 2005; 15: 137-41. [ Links ]
19. Estrada M, Gutiérrez A, Palacios B, Pérez G, Rovira A, Vives J. Estudio bioquímico y molecular de la glucosa-6-fosfato deshidrogenasa en Cuba. Rev Cubana Hematol Inmunol Hemoter. 1995; 15: 1-5. [ Links ]
20. Guindo A, Fairhurst R, Doumbo O, Wellems T, Diallo D. X-linked G6PD deficiency protects hemizygous males but not heterozygous females against severe malaria. PLOS Med. 2007; 4: 516-21. [ Links ]
21. Pinna A, Carru C, Solinas G, Zinellu A, Cata F. Glucose-6-phosphate dehydrogenase deficiency in retinal vein occlusion. Invest Ophthalmol Vis Sci. 2007; 48: 2747-52. [ Links ]
22. Minucci A, Concolino P, Antenucci M, Santonocito C, Ameglio F, Zuppi C, et al. Description of a novel missense mutation of glucose-6-phosphate dehydrogenase gene associated with asymptomatic high enzyme deficiency. Clin Biochem. 2007; 40: 851-5. [ Links ]
23. Pietrapertosa A, Palma A, Campanale D, Delios G, Vitucci A, Tannoia N. Genotype and phenotype correlation in glucose-6-phosphate dehydrogenase deficiency. Haematologica. 2001; 8: 30-5. [ Links ]
24. Filosa S, Giacometti N, Wangwei C, De Mattia D, Pagnini D, Alfinito F, et al. Somatic-cell selection is a major determinant of the blood-cell phenotype in heterozygotes for glucose-6-phosphate dehydrogenase mutations causing severe enzyme deficiency. Am J Hum Genet. 1996; 59: 887-95. [ Links ]
25. Kaplan M, Hammermman C. Onset of Jaundice in G-6-PD deficit neonates. Indian Pediatr. 2006; 43: 459-60. [ Links ]
26. Longo L, Vanegas OC, Patel M, Rosti V, Li H, Waka J, et al. Maternally transmitted severe glucose 6-phosphate dehydrogenase deficiency is an embryonic lethal. EMBO J. 2002; 21: 4229-39. [ Links ]
27. Gonzáles A, Rafael D, Zuñiga H, Carranza E. Actividad especifica de la glucosa-6-fosfato-deshidrogenasa en tejido adiposo y hepático durante la exposición a la altura. Cienc Investig. 1999; 2: 64-8. [ Links ]
28. Weimer TA, Salzano FM, Westwood B, Beutler E. G6PD variants in three South American ethnic groups: population distribution and description of two new mutations. Hum Hered. 1998; 48: 92-6. [ Links ]
29. Chuan SD, Ren X, Chen L, Jiang W, Ha Y, Yang M. Detection of the most common G6PD gene mutations in Chinese using amplification refractory mutation system. Hum Hered. 1999; 49: 133-8. [ Links ]
30. Dal Borgo P, Silva R, Caveires M. Dos nuevas mutaciones de glucose 6 fosfato deshidrogenasa G6PD Santiago y G6PD Calvo Mackenna. Rev Chil Pediatr. 2000; 71: 1-6. [ Links ]

