










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombia Médica
On-line version ISSN 1657-9534
Colomb. Med. vol.40 no.3 Cali July/Sept. 2009
La farmacogenómica en medicina
Carlos Isaza, MD1, Juan C. Sepúlveda-Arias, MD, PhD2, Julieta Henao, MD2
1. Director, Grupo de Investigación en Farmacogenética, Facultad de Ciencias de la Salud, Universidad Tecnológica de Pereira, Pereira, Colombia. e-mail: caisaza@utp.edu.co
2. Miembro del Grupo de Investigación en Farmacogenética, Facultad de Ciencias de la Salud, Universidad Tecnológica de Pereira. e-mail: jcsepulv@utp.edu.co julietahenao@utp.edu.co
Recibido para publicación abril 30, 2009 Aceptado para publicación julio 1, 2009
RESUMEN
Introducción: La farmacogenómica estudia la forma como las variaciones del genoma influyen en la respuesta a medicamentos. Su principal valor médico consiste en: i) la identificación de individuos en quienes se puede predecir si un fármaco será eficaz y a qué dosis o, por el contrario, si el fármaco se debe evitar por alto riesgo de toxicidad o porque el paciente nunca responderá a él; ii) identificar blancos moleculares susceptibles de ser intervenidos por fármacos.
Objetivo: Revisar y sistematizar información farmacogenómica relacionada con la utilización y el impacto clínico de medicamentos.
Metodología: Se consultó la literatura biomédica pertinente en las bases de datos Medline, Proquest, Science Direct, Ovid y Cochrane, así como la información disponible en sitios web de organismos sanitarios internacionales.
Palabras clave: Farmacogenética; Farmacogenómica.
Pharmacogenomics in medicine
SUMMARY
Introduction: Pharmacogenomics studies how changes in the genome influence the response to drugs. Its main medical value consists in: i) the identification of individuals in which it is possible to predict if certain drug and dose will be effective or on the contrary, if the drug must be avoided due to its high toxicity risk or because the patient will never respond to it; ii) to identify molecular targets able to be intervened by drugs.
Objective: To carry out a systematic review about pharmacogenomic oriented to the use and clinical impact of drugs.
Methodology: The pertinent biomedical literature was searched in several databases such as Medline, Proquest, Science Direct, Ovid and Cochrane, as well as the available information in web sites of international sanitary organizations.
Keywords: Pharmacogenetics; Pharmacogenomics.
PRESCRIPCIÓN POR ENSAYO Y ERROR
Una constante de la farmacoterapia es la forma variable como las personas responden a los medicamentos. En efecto, siempre que se emplean fármacos en grupos humanos se encuentran individuos que responden de la manera esperada, otros con falla terapéutica y en algunos los efectos indeseables superan los beneficios. Aun quienes responden en la forma buscada se distribuyen normalmente en relación con las dosis, con personas que requieren dosis usuales de medicamentos, pero otras que responden con dosis más bajas o más altas. Y este fenómeno no se limita a la práctica clínica, también en los ensayos clínicos rigurosos y bien controlados resultan pacientes que responden al tratamiento, otros con respuesta excesiva o insuficiente y en otros predominan los efectos indeseables.
Al reconocer las variaciones individuales en la expresión de enfermedades y en la respuesta a fármacos, el proceso de utilización de medicamentos debe seguir una secuencia racional: basados en la mejor evidencia científica del momento y en el arsenal de medicamentos disponible para determinada enfermedad, grupos de expertos establecen por consenso protocolos de tratamiento. El prescriptor se encarga de individualizar el tratamiento y elegir el fármaco y la dosis que piensa son adecuados, tomando en consideración una serie de variables del paciente y su entorno, relativamente fáciles de visualizar, como su edad, género, peso, comorbilidad, comedicación y condición socio-económica; además, el prescriptor puede recurrir a exámenes paraclínicos a fin de reunir más elementos de juicio. En el contexto clínico esta estrategia brinda la máxima probabilidad de beneficio con la mínima probabilidad de daño en cada caso particular. Por último, en la medida que aparecen los efectos del fármaco se van haciendo ajustes que mejoren su relación riesgo/beneficio, hasta alcanzar la llamada «dosis efectiva individual» (Figura 1).
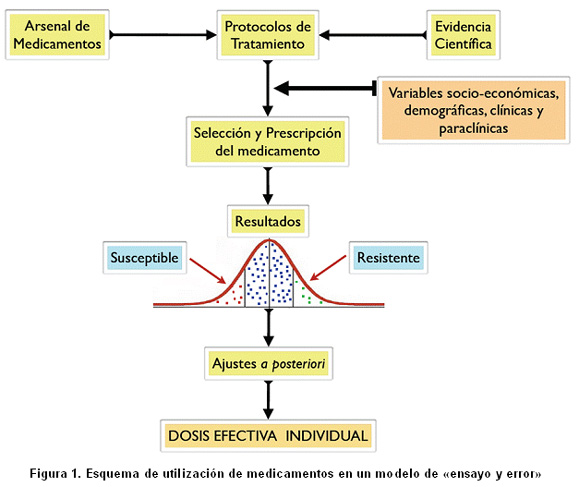
La mayoría de los fármacos sometidos a ajustes de las dosis a priori, de acuerdo con características del paciente, y ajustes a posteriori, con base en la respuesta del paciente, exhiben un aceptable margen de seguridad. En algunos casos, sin embargo, la estrategia de «ensayo y error» puede entrañar riesgos inaceptables y el ajuste empírico de las dosis resultar peligroso e impreciso. En estas circunstancias es deseable incorporar en el acto médico nuevas variables que aumenten el valor predictivo de la formulación y nos acerquen más a la identificación anticipada de las personas que se beneficiarán o no de un tratamiento.
GENÓMICA Y MEDICAMENTOS
Entre las variables históricamente ausentes al tomar decisiones terapéuticas, las relacionadas con los genes pueden llegar a tener el mayor peso en el resultado de un tratamiento. La demostración de que la genética juega un papel en la respuesta a fármacos ha avanzado en forma vertiginosa en dos sentidos:
Heterogeneidad genética de los pacientes. Dentro de la gran identidad de especie, cada ser humano es genéticamente único y está dotado de variantes genéticas que lo diferencian de los demás. La impronta genética de cada individuo determina su forma como se relaciona con los fármacos: la velocidad y la magnitud con que los absorbe, distribuye y elimina, así como la intensidad y el tipo de respuesta de su organismo al medicamento1.
Podría decirse que la sumatoria de las variantes genéticas es lo que hace a cada individuo un ser único e irrepetible. Desde el punto de vista evolutivo, tales diferencias son una seguridad biológica porque funcionan como reserva de supervivencia, en la medida que facilitan la adaptación de la especie en su conjunto a un entorno cambiante. Si la comunidad se expone a un agente agresor de gran impacto sobreviven los individuos genéticamente resistentes; recordemos las grandes epidemias de la Edad Media, las cuales desaparecían tan fácil como irrumpían, cuando mataban a los susceptibles y sobrevivían los resistentes2.
Heterogeneidad genética de la enfermedad. Cada vez es mayor la evidencia con respecto a:
1. Que prácticamente todas las enfermedades caracterizadas actualmente como entidades únicas, realmente son conjuntos de subtipos de la enfermedad que comparten rasgos clínicos, paraclínicos y hasta histopatológicos, pero que se diferencian a nivel molecular, dependiendo de los genes que se expresan o dejan de expresar en cada subtipo. De la enfermedad conceptualizada a nivel de células, órganos y sistemas, se ha pasado a la enfermedad caracterizada en términos de moléculas y genes, y es el patrón genético expresado el que determina en últimas el éxito o el fracaso de un tratamiento3. A modo de ejemplo, las pacientes her-2 positivas representan una subcategoría (en términos de pronóstico y respuesta al tratamiento) de las pacientes con un diagnóstico más amplio llamado «cáncer de mama».
2. Que prácticamente todas las enfermedades comunes son de naturaleza multifactorial, fruto de la concurrencia de factores genéticos y ambientales, con importancia relativa de cada uno de ellos, de tal forma que en algunas enfermedades los factores externos parecen más importantes, mientras en otras priman los factores internos.
FARMACOGENÉTICA
El polimorfismo es una variación en la secuencia del ADN que se encuentra en más del 1% de los individuos de una población. Los polimorfismos comprenden las sustituciones de una sola base, donde un solo nucleótido (A, C, G ó T) es reemplazado por otro (single nucleotide polymorphisms: SNPs), deleciones o inserciones de bases (deletion insertion polymorphisms: DIPs) y variaciones repetidas como microsatélites (short tandem repeats: STRs). Los SNPs dan cuenta de alrededor de 90% de la variación y se encuentran dispersos por todo el genoma humano4.
La búsqueda del impacto de las variaciones del genoma humano en la respuesta a los fármacos se expandió en los últimos años gracias a la culminación exitosa del Proyecto Genoma Humano y, más recientemente, del Proyecto HapMap Internacional5, el cual define patrones de asociación entre diferentes variantes génicas y permite seleccionar un mínimo de SNPs que capturen la máxima diversidad del genoma humano; el uso de tales SNPs evita tener que genotipificar todos los alelos4. Por supuesto, no se puede ignorar el acompañamiento de las poderosas herramientas de la bioinformática, la biotecnología y las técnicas experimentales disponibles; con cuya ayuda se ha hecho cada vez más accesible la información contenida en el genoma humano6,7.
Hay múltiples mecanismos por los cuales un polimorfismo resulta en un fenotipo alterado de respuesta a un fármaco:
1. Cambia la secuencia de aminoácidos de la proteína, lo que da como resultado disminución, pérdida o incremento de su función (por ejemplo, se modifica la afinidad de un receptor o la actividad de una enzima por el fármaco).
2. Se altera la región del promotor de un gen, modificando su transcripción y la consiguiente cantidad de proteína expresada.
3. Se pierde el gen o, por el contrario, se producen varias copias de él, lo que se traduce en ausencia o excesivas cantidades de enzima y, en consecuencia, el portador será un metabolizador lento o ultra-rápido de los fármacos sustratos de la enzima7.
Un biomarcador genómico se define como «una característica del ADN o del ARN indicadora de procesos biológicos normales, patogénicos o de respuesta a una intervención»8. De la interfase entre genética y farmacología se ha originado esta nueva disciplina llamada farmacogenómica, la cual ha sido definida por el Centro para la Evaluación e Investigación de Fármacos (CDER) de la FDA como «la investigación de las variaciones del ADN y del ARN relacionadas con respuesta a fármacos»; una subcategoría de ella es la farmacogenética, definida como «la influencia de las variaciones del ADN en la respuesta a fármacos» (http://www.fda.gov/cder/guidance/).
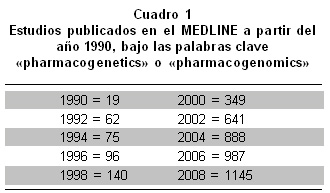
Como la mayoría de autores está de acuerdo en considerar los términos farmacogenómica y farmacogenética como sinónimos, en este artículo se usarán en forma indistinta. Para poner las cosas en una perspectiva histórica, en el Cuadro 1 se muestran los datos de los estudios publicados en MedLine en los últimos 10 años bajo las palabras claves «pharmacogenetics» y «pharmacogenomics». El paso de unas cuantas a centenares de publicaciones científicas en el curso de unos pocos años demuestra el interés de la comunidad científica por esta disciplina en expansión, aunque hay que aceptar que sigue siendo un tema relativamente marginal, si se compara con otros como la hipertensión, la diabetes y la falla cardíaca, que están en el orden de 5 a 10 mil artículos publicados por año.
Tradicionalmente los estudios farmacogenéticos han empezado con el descubrimiento de un efecto indeseable relevante o de una amplia variabilidad en los efectos de un fármaco, y continúan con la búsqueda de la base genética de esa respuesta. En el ejemplo más sencillo, a individuos que no responden o lo hacen en forma exagerada a un fármaco, se les miden concentraciones sanguíneas del mismo y se encuentra que varían ampliamente en comparación con quienes responden de la manera habitual a dosis similares; enseguida se investiga la ruta metabólica del fármaco y se halla que la enzima responsable de su metabolismo funciona de manera defectuosa o se encuentra en cantidades inusuales (fenotipo); finalmente el análisis del gen que codifica la enzima revela variaciones que explican su cantidad o funcionamiento anormal (genotipo)6.
Con el «dogma de la biología» (el ADN se transcribe en ARN, el ARN se traduce en proteínas, las proteínas participan en procesos biológicos) como telón de fondo, la farmacogenética partió de la premisa de que la estructura genética del individuo tiene un papel determinante en la respuesta a fármacos y, por tanto, era posible explicar una respuesta farmacológica a partir de un genotipo. En los primeros años de su desarrollo, los estudios farmacogenéticos se enfocaron en los genes involucrados en procesos farmacocinéticos, especialmente el metabolismo y transporte de fármacos a través de membranas biológicas. Como la respuesta farmacológica corresponde a un fenotipo complejo en el que también están implicados genes que participan en la secuencia de eventos que van desde el momento en que el fármaco interacciona con su receptor hasta la aparición de los efectos terapéuticos o tóxicos, rápidamente la búsqueda de marcadores farmacogenómicos se extendió a todos los procesos biológicos que se dan a partir del momento en que un fármaco y un organismo entran en contacto5.
Los fármacogenes asociados con seguridad o eficacia terapéutica (Pharmacogenetics Research Network: http://www.nigms.nih.gov/Initiatives/PGRN/) pueden clasificarse en cuatro categorías:
1. Farmacocinéticos. Relacionados con la absorción, distribución, metabolismo o excreción de fármacos.
2. Farmacodinámicos. Implicados en el mecanismo de acción y efectos de los fármacos. Se incluyen los genes que codifican receptores de fármacos y proteínas funcionales involucradas en los eventos post-receptor. Los polimorfismos de estos dos grupos de genes suelen ser neutrales, no confieren ventajas ni desventajas y sus consecuencias fenotípicas se visualizan sólo cuando el individuo se expone al fármaco.
3. Modificadores de enfermedad. Son genes del paciente comprometidos a la vez con una enfermedad y con una respuesta farmacológica. Por ejemplo, algunos polimorfismos de canales iónicos predisponen al paciente a arritmias cardíacas (las llamadas «canalopatías»), las cuales pueden ser precipitadas por medicamentos que prolongan el intervalo QT; en este caso la misma variante alélica predispone al paciente a enfermedad y a toxicidad farmacológica.
4. Genes de procesos neoplásicos que funcionan como marcadores de respuesta a fármacos, como el oncogen her-2 del cáncer de mama7.
Podría esperarse la existencia de una quinta categoría de polimorfismos genéticos con funciones biológicas que se asemejen a las de fármacos y protejan o sirvan para tratar enfermedades; es decir, verdaderos genes-fármacos denominados por algunos como «genes farmacomiméticos». Un buen indicio de la existencia de variantes genéticas farmacomiméticas está en la reciente descripción de un polimorfismo del gen GRK5 (quinasa 5 acoplada a receptor de proteína-G), que se comporta de manera similar a lo que ocurre cuando se bloquea el receptor adrenérgico b-1 en pacientes con falla cardíaca. Este descubrimiento tuvo su origen en la búsqueda de las razones por las cuales son tan variables las respuestas de los pacientes afroamericanos al tratamiento de la insuficiencia cardíaca crónica con agentes beta-bloqueadores. Se encontró que los enfermos afroamericanos portadores del alelo farmacomimético denominado GRK5L41 tenían la misma protección contra muerte y trasplante cardíaco que los pacientes que tomaban beta-bloqueadores9; en otras palabras, el alelo GRK5L41 y los fármacos beta-bloqueadores son equiefectivos en afroamericanos con falla cardáaca. En principio sería interesante determinar la prevalencia de este polimorfismo en otros grupos étnicos y definir si su efecto protector se conserva entre no-afroamericanos.
DIFERENCIAS ÉTNICAS
Existen grandes diferencias en las frecuencias con que se presentan ciertas respuestas a fármacos entre distintos grupos étnicos. A principios de la década de 1980 ya se habían informado diferencias étnicas en la respuesta a los fármacos antihipertensivos y desde el llamado «cuarto JOINT sobre hipertensión» (JNC-IV), publicado en 1988, se recomendaba considerar la etnicidad en la selección del agente antihipertensivo. Actualmente ésta es una variable considerada de rutina en los protocolos de tratamiento de la hipertensión10. Sin embargo, a fin de evitar generalizaciones inadecuadas, se debe reconocer que se presentan diferencias étnicas en la respuesta a fármacos cuando también existen diferencias en las frecuencias con que se presentan los polimorfismos genéticos involucrados en los efectos farmacológicos; es decir, que el verdadero predictor de respuesta farmacológica es el genotipo y no la etnicidad y que la variable farmacogenética aporta más que la etnicidad a un resultado farmacológico10.
La warfarina, el anticoagulante oral más empleado en el mundo, representa un buen ejemplo de diferencias étni-cas en su efecto, porque las dosis promedio de warfarina para alcanzar un INR entre 2 y 3 son de 3.4 mg/día en orientales, 5.1 mg/día en caucásicos y 6.1 mg/día en negros. Como se muestra en el Cuadro 2, tales hallazgos se correlacionan muy bien con las prevalencias de alelos de susceptibilidad a la warfarina en los dos genes implicados en el metabolismo (CYP2C9) y en el sitio de acción del fármaco (VKORC1); la mayor prevalencia de metabolizadores lentos del fármaco en caucásicos podría explicar las diferencias de dosis con los negros, mientras que la alta prevalencia en población oriental de alelos de susceptibilidad en el gen VKORC1 puede explicar cómodamente la alta sensibilidad de este grupo étnico a la warfarina, en comparación con caucásicos y negros10.

Todavía más interesante resulta el caso del bucindolol, un agente beta-bloqueador y vasodilatador desarrollado para el tratamiento de la insuficiencia cardíaca crónica, que está a punto de convertirse en el primer fármaco cardiovascular en ser aprobado con un uso genéticamente determinado8. A diferencia de otros beta-bloqueadores, inicialmente no se pudo demostrar disminución de la mortalidad con este agente; luego se encontró que los beneficios del fármaco en la falla cardíaca dependían del grupo étnico y que mientras en los negros no mostraba eficacia, reducía de forma significativa la mortalidad en personas no-negras. Finalmente, en una serie de investigaciones interesantes se encontró que, cuando las poblaciones estudiadas se estratificaron por el genotipo Arg389Gly del gen del receptor adrenérgico beta-1 (ADRB1) y el genotipo Ins/Del del gen ADRA2C (codifica un subtipo del receptor adrenérgico b-2), los pacientes homocigotos Arg389Arg o Ins/Ins tuvieron reducción significativa de las tasas de hospitalización y mortalidad, en comparación con los no portadores de estos alelos. Las investigaciones concluyen que, a diferencia de los demás bloqueantes beta, el bucindolol es un fármaco con dos propiedades farmacogenéticas únicas: los pacientes con falla cardíaca portadores del genotipo Arg389Arg del gen ADRB1 o del genotipo Ins/Ins del gen ADRA2C son los beneficiados por el medicamento, con el máximo beneficio conseguido en quienes portan ambos genotipos. Como las prevalencias de ambos genotipos Arg389Arg e Ins/Ins son menores en negros (32% y 38%) que en blancos (55% y 87%), eran de esperarse diferencias étnicas en los beneficios proporcionados por el bucindolol en pacientes con insuficiencia cardíaca. Sin embargo, es claro que tales diferencias se deben a factores genéticos y así como hay negros con genotipo «respondedor al bucindolol», también hay blancos con genotipo «no respondedor»10.
BIOMARCADORES FARMACOGENÓMICOS
No se sabe cuántos genes quedan implicados a partir del momento en que un fármaco y un organismo humano se ponen en contacto, pero sí se sabe que el perfil genético del individuo permanece estable a lo largo de la vida, a diferencia de otras variables demográficas, clínicas y medioambientales influyentes en respuestas farmacológicas1. El proyecto PharmGKB (The Pharmacogenetics and Pharmacogenomics Knowledge Base, http://www.pharmgkb.org/) reconoce hasta ahora 182 y 937 genes asociados, respectivamente, con la farmacocinética y con la farmacodinamia; además el proyecto describe 39 «fármacogenes muy importantes» (VIP, por su sigla en inglés) que corresponden a los genes de particular relevancia actual en farmacogenómica (Cuadro 3).
A continuación se revisan algunos ejemplos de marcadores farmacogenómicos bien caracterizados, divididos entre aquellos que inciden sobre las propiedades farmacocinéticas del medicamento (absorción, distribución, metabolismo y excreción), los que afectan sus propiedades farmacodinámicas (mecanismo de acción, cascada de eventos post-receptor y efectos) y marcadores tumorales.
Biomarcadores que inciden en la farmacocinética
1. Enzimas metabolizadoras de fármacos. Se acepta que las enzimas capaces de degradar químicos a los cuales se exponen los organismos vivos aparecieron como un fenómeno de co-evolución entre plantas y herbívoros. Las células animales se fueron armando así de todo un arsenal de enzimas capaces de inactivar sustancias químicas exógenas, conocidas como xenobióticos. Esta estrategia resultó biológicamente tan exitosa que se extendió a todos los seres vivos y ahora hace parte de los principios que gobiernan las interacciones entre sistemas biológicos y sustancias químicas, son un componente de la dinámica de la vida y de la muerte2,11.
Por hacer parte de la primera línea de defensa para evitar el ingreso de sustancias exógenas potencialmente nocivas al interior del organismo, las enzimas metabolizadoras de fármacos, que forman parte de las enzimas xenobióticas, exhiben algunas características destacables. La primera es su amplia especificidad de sustrato, porque cada una de ellas es capaz de metabolizar muchos fármacos, así como un mismo fármaco puede ser metabolizado por varias enzimas, aunque siempre habrá una ruta metabólica principal para cada fármaco. En segundo lugar, la mayoría de ellas son fácilmente inducibles o inhibibles por los propios xenobióticos, los cuales pueden competir entre sí por la misma enzima12. Por último, existe un alto grado de polimorfismo genético en muchas de ellas, que da origen a los distintos fenotipos hallados en la población: la mayoría de los individuos tiene actividad enzimática normal y se clasifica en el fenotipo «metabolizador eficiente» (EM); algunas personas pueden heredar variantes alélicas que codifican enzimas con actividad catalítica deficiente o nula (fenotipo «metabolizador pobre o lento», PM); en casos puntuales también se encuentran individuos con mutaciones o varias copias funcionales de un gen, capaces de expresar isoenzimas más activas o cantidades excesivas de enzima (fenotipo «metabolizador ultrarrápido», UM)7.
Estas características de las enzimas xenobióticas resultan importantes para la supervivencia de la especie. Por un lado, la capacidad de las enzimas para ser inhibidas o inducidas y las relaciones promiscuas entre enzimas y sustratos facilitan la detoxificación y le permite al organismo adaptarse a los cambios de su entorno químico; con algunos fármacos, por ejemplo, si se eleva su concentración también se eleva la actividad de la enzima que lo degrada. Por otro lado, los polimorfismos de los genes que codifican estas enzimas son prenda de garantía de supervivencia de la especie en la medida en que, como ya se dijo, si la comunidad se expone a un tóxico de gran impacto, sobrevivirán los individuos portadores de las variantes genéticas que confieren resistencia a la agresión13,14.
Se debe tener presente que los profármacos son metabolizados al compuesto activo en el cuerpo del paciente, de modo que las personas deficitarias en la vía activante del fármaco tienen mínimo o ningún beneficio al consumirlo. Con profármacos como la codeína, las consecuencias de esta carencia no son graves y pueden ser fácilmente manejadas, pero con otros profármacos, como el clopidogrel o el tamoxifen, las consecuencias pueden ser fatales en pacientes con enfermedad coronaria o cáncer de mama, respectivamente8.
Enzimas del citocromo P-450. A mediados del siglo pasado se descubrieron en hepatocitos acúmulos de pigmentos que absorben la luz a 450 nm, por lo que se les denominó citocromos P-450 (CYP). Luego se esclareció que tales pigmentos correspondían a un enorme grupo de enzimas con similitudes estructurales entre sí, razón por la cual fueron clasificadas como una superfamilia. Las enzimas de una misma familia (designadas por un número arábigo: CYP1, CYP2, CYP3) tienen una homología en la secuencia de aminoácidos no menor de 40%; cada familia se divide en subfamilias (designadas por una letra: CYP1A, CYP2D, CYP3A) con una homología mayor de 77% en su secuencia de aminoácidos. Cada enzima específica se designa por un segundo número arábigo: CYP1A2, CYP2D6, CYP3A4. Por convención, cuando se hace referencia al gen que codifica la enzima se emplea la misma nominación, pero en letra itálica: CYP1A2, CYP2D6, CYP3A4. Cada una de las variantes o alelos del mismo gen se representa con una letra mayúscula, separada del correspondiente gen por un asterisco; por ejemplo, los alelos CYP2D6*3 y CYP2D6*4. Para la nomenclatura de los alelos CYP se puede consultar el sitio http://www.cypalleles.ki.se15.
En humanos se han descrito al menos 18 familias y 44 subfamilias CYP metabolizadoras de xenobióticos, de las cuales sólo las familias CYP1, CYP2 y CYP3 tienen importancia en el metabolismo de fármacos (Cuadro 4)7,16. Recientemente se revisó la ruta de eliminación de los 200 medicamentos más vendidos por prescripción en los EEUU y se encontró que cerca de 80% de los fármacos son metabolizados por las familias 1, 2 y 3 del CYP-450 y que la mayor contribución la hacen las isoenzimas CYP3A4/5 (37%), CYP2C9 (17%), CYP2D6 (15%), CYP2C19 (10%), CYP1A2 (9%), CYP2C8 (6%) y CYP2B6 (4%). Las enzimas CYP1A2, CYP2C8 y CYP3A4, que carecen de polimorfismos funcionales, son responsables del metabolismo de la mitad de estos fármacos, mientras la otra mitad se metaboliza por la ruta de las isoenzimas CYP2B6, CYP2C9, CYP2C19 y CYP2D6, cuyos genes son ricos en polimorfismos que causan cambios en la expresión, selectividad o actividad de la enzima, que se reflejan en variabilidad en la respuesta a fármacos17.
CYP2B6. Codificada por uno de los genes CYP más polimórficos, con más de 100 variaciones descritas. Se calcula que la enzima CYP2B6 da cuenta entre 3% y 6% del pool microsomal hepático pero, debido a los polimorfismos genéticos, hay variabilidad interindividual de hasta 100 veces en los niveles hepáticos de la enzima; por ejemplo, la variante alélica más común (CYP2B6*6) reduce hasta en 75% la expresión de la enzima. Ejemplos farmacogenéticos: neurotoxicidad por efavirenz y cardiotoxicidad por metadona (síndrome de QT largo) en homocigotos mutados 2B6*6, pertenecientes al fenotipo «metabolizador lento»17.
CYP2C9. La enzima se expresa abundantemente en el hígado, es genéticamente polimórfica y metaboliza algunos medicamentos con estrecho margen terapéutico. Los alelos *2 y *3 son los más estudiados y se relacionan con disminución de hasta 90% de la actividad de la enzima, dependiendo del fármaco sustrato. Ejemplos farmacogenéticos: en metabolizadores lentos hay mayor incidencia de hipoglicemia por hipoglicemiantes orales, de gastropatía por AINEs y de sangrado por warfarina17.
CYP2C19. De los cuatro genes de la subfamilia CYP2C, el gen de la isoenzima 2C19 fue el primero en el que se identificaron alelos nulos asociados con el fenotipo «pobre metabolizador» (PM). Ha sido objeto de mucha investigación farmacogenética no sólo por tener entre sus sustratos agentes tan importantes como los antiulcerosos inhibidores de la bomba de protones (iBP), el antiagregante plaquetario clopidogrel y algunos antidepresivos de primera línea, sino porque existen grandes diferencias en las frecuencias de «pobres metabolizadores» entre los grupos étnicos: 1%-3% de mestizos18, 5% de blancos y negros y hasta 20% de orientales. Ejemplos farmacogenéticos: las personas pertenecientes al fenotipo EM metabolizan los iBP a una velocidad tal que requieren dosis hasta cuatro veces mayores que los individuos con fenotipo PM, para alcanzar concentraciones séricas y efectos similares del fármaco19; los individuos con el fenotipo PM tienen menor efecto antiplaquetario con clopidogrel, en razón a que éste es un profármaco que debe ser activado por esta enzima17.
CYP2D6. Debido a que se han identificado numerosas variantes del gen CYP2D6, que van desde la carencia absoluta del gen hasta su multiplicación, pasando por una gran cantidad de SNPs deficientes, todas las poblaciones están estratificadas fenotípicamente en metabolizadores lentos (PM), eficientes (EM) y ultra-rápidos (UM) de aproximadamente 20% de los medicamentos que consume el ser humano y que son metabolizados (activados o inactivados) por esta ruta. Existen diferencias en las frecuencias con que se distribuyen los diferentes fenotipos CYP2D6 entre los grupos étnicos; por ejemplo, entre 7% y 10% de caucásicos, 7% de mestizos, 4% de afroamericanos y sólo entre 1% y 2% de los orientales y africanos corresponden al fenotipo PM20.
Con respecto al fenotipo UM, hasta ahora se ha hallado la presencia entre 2 y 13 copias funcionales del gen. Igual que ocurre con los otros fenotipos, las frecuencias de UM son diferentes entre los grupos étnicos. Se han informado frecuencias de UM entre 20% y 29% de algunas poblaciones africanas, 7%-10% de españoles, 2% de mestizos y 1% de caucásicos. En relación con el origen de la multiplicación del gen se ha formulado la hipótesis de que la más alta prevalencia informada en africanos, seguida por españoles y después por mestizos americanos se podría explicar porque este alelo tuvo origen en África, fue transmitido a los españoles durante la migración musulmana a la Península Ibérica, y ellos lo transmitieron a los mestizos a partir del descubrimiento de América20.
El impacto farmacológico de los polimorfismos CYP2D6 ha sido explorado con un amplio número de fármacos y es difícil entender por qué la genotipificación CYP2D6 aún no se usa en la práctica médica, si se tiene en cuenta que la efectividad y la tolerabilidad de muchos medicamentos dependen de la actividad de esta enzima. Se calcula que la genotipificación predictiva CYP2D6 podría ser benéfica entre 30% y 40% de los fármacos sustratos, aunque todavía faltan estudios clínicos prospectivos confirmatorios21. Con respecto a la inquietud de si las pruebas farmacogenéticas son costo-efectivas, en un estudio conducido en los EU se encontró que los problemas generados por psicofármacos sustratos de la enzima CYP2D6 en pacientes psiquiátricos metabolizadores lentos y ultra-rápidos imponen un costo adicional entre 4,000 y 6,000 dólares por paciente/año22. De otra parte, resultó muy importante demostrar que el tamoxifen, un profármaco cuyo metabolito activo (endoxifen) se genera por la vía CYP2D6, se asocia con menor tiempo de recurrencia y menor sobrevida en mujeres con cáncer de mama y fenotipo PM8.
Subfamilia CYP3A. Las isoenzimas 3A4 y 3A5 contribuyen al metabolismo de la mayor cantidad y más variados grupos de medicamentos de consumo por el ser humano, aunque la 3A5 se expresa mucho menos que la 3A4 y carece de sustratos específicos. Estas enzimas están localizadas en órganos de particular relevancia en la disposición de fármacos (TGI, hígado y riñón) y poseen mecanismos de regulación complejos; por un lado, muchos fármacos actúan como ligandos de «receptores nucleares huérfanos», que a su vez regulan la expresión de los genes CYP3A4/5, dando como resultado una muy fácil inhibición o inducción de estas enzimas. Por otro lado, la CYP3A4 es la única enzima del citocromo P-450 que muestra diferencias de género y se expresa hasta dos veces más en mujeres que en hombres. La facilidad con que la actividad enzimática puede ser modulada contrasta con el hecho de que no se han demostrado correlaciones genotipo-fenotipo farmacológico y no existe evidencia de una contribución significativa de los polimorfismos genéticos en la actividad de la enzima. Otra interesante característica de esta enzima es que funciona en forma concertada con la glicoproteína P (Gp-P) para reducir la concentración intracelular de xenobióticos17.
A diferencia de las enzimas que exhiben polimorfismo genético, con las cuales es relativamente sencillo hacer la caracterización de los individuos mediante pruebas de genotipificación altamente confiables, la evaluación de la actividad catalítica de la enzima CYP3A tiene complicaciones especiales. Tal circunstancia ha ocasionado trágicas sorpresas como las de astemizol, terfenadina y cisaprida, metabolizados vía CYP3A4, pero cuyo metabolismo resultó fácilmente bloqueable por un numeroso grupo de fármacos (Cuadro 4), acumulándose los compuestos originales a niveles cardiotóxicos que resultaron letales en varios casos7.
N-acetiltransferasa tipo 2 (NAT2). La acetilación es una de las rutas metabólicas más activas en la degradación de xenobióticos. Varios alelos del gen NAT2 se traducen en una enzima de baja actividad, dividiendo a la población en acetiladores «rápidos» (AR) y «lentos» (AL) de fármacos como isoniacida, hidralazina, dapsona, sulfas, dipirona y cafeína. En negros y población caucásica de Europa y Norte América hay alrededor de 70% de acetiladores lentos, mientras en las poblaciones orientales corresponden sólo entre 10% y 30%. Los hispanos aparecen en un lugar intermedio entre blanco/africanos y asiáticos con 60% de AL23. Aunque no se han establecido en forma concluyente las consecuencias clínicas del fenotipo acetilador en el metabolismo de fármacos, sí se ha asociado el fenotipo AL con mayor riesgo de neuropatía por isoniacida, de síndrome lúpico inducido por hidralazina y de reacciones tóxicas provocadas por sulfas. Sin embargo, aunque este marcador farmacogenético se conoce desde hace 50 años, la caracterización genotípica NAT2 nunca hizo ingreso a la práctica clínica.
Metiltransferasas. La metilación es una importante vía del metabolismo de fármacos, hormonas, neurotransmisores y macromoléculas como proteínas, ARN y ADN. La tiopurina S-metiltransferasa (TPMT), una enzima genéticamente polimórfica, cataliza la metilación de los fármacos del grupo de las tiopurinas (azatioprina, mercaptopurina y tioguanina). La actividad de TPMT varía entre diferentes grupos étnicos24, y la farmacogenética de la TPMT representa uno de los mejores ejemplos del potencial de implicaciones clínicas del polimorfismo genético de una enzima metabolizante de fármacos, porque las tiopurinas tienen relativamente estrecho índice terapéutico y además se usan para tratar situaciones que ponen en peligro la vida, tales como leucemia linfoblástica aguda, enfermedades autoinmunes, o en personas que requieren trasplantes de órganos23.
Usuarios de tiopurinas con baja o ausente actividad TPMT están en riesgo de sufrir mielosupresión inducida por estos fármacos. Debido a las consecuencias potencialmente catastróficas de los polimorfismos deficientes se recomienda la genotipificación en potenciales usuarios. Sin embargo, en el año 2005 sólo en 12% de los departamentos de oncología, hematología y pediatría de los EEUU aplicaban con regularidad la geno o la fenotipificación antes de administrar tiopurinas25.
Recientemente se demostró una favorable relación de costo-efectividad del genotipo TPMT previo al tratamiento con tiopurinas en niños con diagnóstico de leucemia linfoblástica aguda (ALL). Concluyen los autores que la genotipificación TPMT debería de ser seriamente considerada como parte integral de la atención previa al tratamiento con tiopurinas25.
2. Transportadores de fármacos. A medida que aumentó la evidencia de que el sólo metabolismo de fármacos no podía dar cuenta de toda la variabilidad en la respuesta a medicamentos, se inició la exploración de otros procesos que también pudieran ser determinantes en la disposición de los fármacos. Los transportadores son proteínas responsables de acarrear moléculas de fármacos a través de las membranas biológicas y, por lo tanto, juegan un papel clave en los procesos de absorción, distribución, metabolismo y excreción de fármacos. La glicoproteína-P (P-gp), por ejemplo, uno de los transportadores mejor caracterizado, se encuentra en sitios estratégicos del organismo y funciona como bomba de eflujo, llevando en forma activa moléculas desde el interior al exterior de células y tejidos, lo que se traduce en cambios de la absorción intestinal, la distribución en el SNC, la excreción biliar y la eliminación urinaria. Sujetos portadores de variantes alélicas defectuosas del gen que codifica la P-gp (llamado gen MDRI) tienen aumentada la absorción de muchos fármacos y a las mismas dosis exhiben concentraciones sanguíneas superiores, comparados con quienes no tienen los polimorfismos del gen26.
Las potenciales consecuencias funcionales de los polimorfismos en los genes que codifican transportadores han sido difíciles de precisar, debido a la complejidad y ubicuidad de todo el sistema de acarreadores de fármacos y a que en muchos casos no se modifican los niveles séricos sino los intracelulares, con las consecuentes limitaciones en el diseño de estudios que exigen determinar la concentración del fármaco en tejidos. Un buen ejemplo es el del metformin, un agente antidiabético que mejora la captación de glucosa en músculo e hígado. El transportador OCT1 (transportador 1 de cationes orgánicos) es el encargado de la captación hepática del fármaco, un prerrequisito para que éste desempeñe su función intracelular. Se ha demostrado que algunos polimorfismos defectuosos del gen OCT1 se asocian con disminución del efecto metabólico del medicamento, debido a la reducción de su acumulación hepática, sin que se vea afectada la concentración sérica del fármaco27. Este estudio ilustra no sólo las dificultades técnicas para demostrar algunos efectos farmacogenéticos en humanos, sino que sus resultados contrarían un paradigma de la farmacología, según el cual existe una relación directa entre niveles sanguíneos de fármaco y magnitud de su efecto, pues en este caso la concentración tisular del medicamento no es reflejo de su concentración sanguínea.
BIOMARCADORES QUE INCIDEN EN LA FARMACODINÁMICA
La información sobre los factores genéticos que afectan el metabolismo y el transporte de fármacos excede en mucho la de los factores que inciden sobre la respuesta. La identificación de los genes y los polimorfismos implicados en los fenotipos de respuesta a fármacos es una labor más ardua, pues la búsqueda con frecuencia debe incluir no sólo las moléculas blanco del fármaco y las que están implicadas en los eventos post-receptor, sino otras vías relacionadas8. Como la mayoría de las veces existe poca información acerca de la ruta de acción del fármaco, por lo general se requieren métodos de búsqueda de grandes porciones del genoma («whole-genome analysis»), técnicas de alto rendimiento y altísimo costo, capaces de detectar SNPs hasta en centenares de miles de segmentos a lo largo del genoma e identificar toda una gama de genes candidatos a estar asociados con la respuesta28. Algunos ejemplos son:
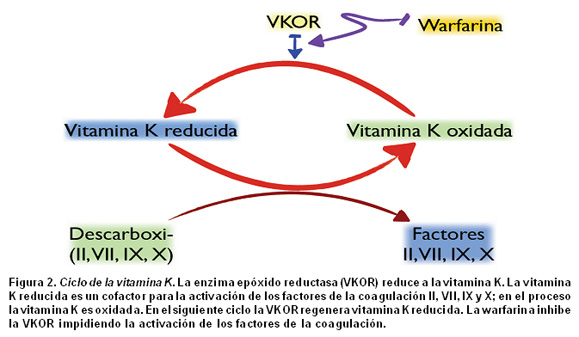
Vitamina K epóxido reductasa (VKOR). La warfarina inhibe esta enzima, codificada por el gen VKORC1, y en esa forma impide la activación de los factores de la coagulación II, VII, IX y X, que dependen de la vitamina K reducida (Figura 2). En las dosis efectivas individuales de warfarina inciden factores genéticos relacionados con los polimorfismos, tanto del gen VKORC1 como del gen que codifica la enzima CYP2C9, cuya función es inactivar el medicamento29,30. De acuerdo con la evidencia disponible, en el año 2007 la FDA le exigió al laboratorio farmacéutico que en el inserto del producto se incluyera la recomendación a los prescriptores para que tuvieran en cuenta los genotipos CYP2C9 y VKORC1, aunque no se mencionó cómo utilizar la información genética en el estimativo de las dosis10. Finalmente, mediante un estudio conducido en usuarios de warfarina de diferentes grupos étnicos, recientemente se validó el algoritmo de dosificación del fármaco basado en variables demográficas, clínicas y farmacogenéticas; de hecho, ya se ha creado el sitio web (www.warfarindosing.org) para ayudar a los clínicos en el cálculo de las dosis del fármaco30.
Receptores adrenérgicos b-2. Ya se ha establecido que las variantes alélicas Arg16Gly y Gln27Glu del gen ADRB2, que codifica para el receptor beta-2 adrenérgico, son marcadores farmacogenéticos. Aunque no todos los estudios concuerdan en los resultados, la mayor evidencia sugiere, por ejemplo, que el alelo Gly16 se asocia no sólo con severidad del asma, sino con poca respuesta a los broncodilatadores agonistas beta-2, en comparación con las personas portadoras del alelo nativo Arg16. De otra parte, los beta-bloqueadores son un grupo de medicamentos con excelente margen de seguridad y amplia gama de efectos terapéuticos, sobre todo en el área cardiovascular. Entre los efectos indeseables de este grupo de agentes se encuentra la dislipidemia, que incluye aumento de triglicéridos y descenso de HDL-C; este es un clásico efecto adverso de tipo farmacogenético, donde el alelo Glu27 del receptor ADRB2 es el biomarcador del riesgo31,32.
Receptor de la vitamina D (VDR). En algunos casos, medicamentos utilizados en el tratamiento de la misma enfermedad pueden tener efectos opuestos según el genotipo del paciente. Nguyen TV et al.33 citan el curioso ejemplo de mujeres con osteoporosis tratadas con alendronado o raloxifeno. Las pacientes con el alelo b del gen VDR tuvieron mayor incremento de la densidad mineral ósea (DMO) que aquellas con la variante B del mismo gen; por el contrario, las pacientes tratadas con raloxifeno y portadoras del alelo B tuvieron mayor incremento de la DMO que las pacientes portadoras del alelo b. En el grupo de mujeres tratadas con ambos fármacos no hubo asociación entre el polimorfismo del gen VDR y los cambios en la DMO.
BIOMARCADORES TUMORALES
Los diferentes perfiles de expresión génica de una enfermedad no sólo pueden arrojar información con respecto al curso natural de la enfermedad (por ejemplo, marcadores tumorales que indican un riesgo incrementado de metástasis) sino también usarse como blancos críticos contra los cuales dirigir «balas» farmacológicas altamente específicas. De hecho el desarrollo de marcadores genómicos relacionados con respuesta al tratamiento es uno de los escenarios más promisorios de la farmacogenómica. Algunos biomarcadores de este tipo ya han sido aprobados por la FDA en la categoría de «pruebas requeridas» (http://www.fda.gov/cder/genomics/genomic_biomarkers_table.htm); estos son algunos ejemplos:
Receptor del factor de crecimiento epidérmico (EGFR). Cetuximab y erlotinib bloquean el EGFR y se usan en algunos tipos de cáncer. Mutaciones activantes del gen EGFR se asocian con respuesta a estos agentes, por lo que resultan útiles únicamente en pacientes con evidencia inmunohistoquímica de sobreexpresión del EGFR.
Receptor 2 de la familia del factor de crecimiento epidérmico (Her2/neu). En el tratamiento de cáncer de mama el trastuzumab, un anticuerpo monoclonal humanizado, ha resultado útil sólo en pacientes que sobreexpresan el receptor Her2/neu en su tejido tumoral. Las pacientes deben ser seleccionadas con este criterio, porque el fármaco es inefectivo en los dos tercios de pacientes que no sobreexpresan el blanco del fármaco.
Cromosoma Filadelfia. En el tratamiento de adultos con leucemia linfoblástica aguda el dasatinib está indicado únicamente en el subgrupo de pacientes con cromosoma Filadelfia positivo (Ph+ALL).
IMPACTO CLÍNICO
Es largo y tortuoso el camino a recorrer para que un marcador farmacogenómico llegue a ser validado y adoptado en la práctica médica. Se debe demostrar primero que el polimorfismo se asocia con un rasgo y se refleja en un fenotipo, es decir, que existe asociación genotipo-fenotipo y tiene real valor predictivo de respuesta farmacológica. Enseguida se debe confirmar, mediante ensayos clínicos controlados y estudios de costo-efectividad, que la genotipificación prospectiva es clínicamente relevante y aporta en forma significativa a la toma de decisiones terapéuticas. Por último, es necesario demostrar que los hallazgos en un grupo étnico pueden ser extrapolados a otro grupo étnico, porque si el verdadero beneficio de una prueba farmacogenética está en encontrar oportunamente los pacientes que se salen de la media y responden a dosis inusualmente altas o bajas, el impacto en cada grupo étnico dependerá de la prevalencia de los polimorfismos responsables de dicha respuesta, es decir, del porcentaje de personas que se beneficiarían de la prueba3,8,34,35. Un estudio reciente en 5,052 usuarios de warfarina con INR entre 2 y 3 validó un algoritmo de dosificación basado en variables demográficas, clínicas y farmacogenéticas al confirmar que, si bien el algoritmo no es mejor que los esquemas convencionales de dosificación de warfarina en 54% de las personas que responden adecuadamente a las llamadas dosis usuales, sí tiene valor predictivo para detectar a 46% de los pacientes que requieren dosis mayores o menores que las medias, por ser portadores de genotipos inusuales30. Los resultados de este estudio, sin embargo, podrían no ser extrapolables a grupos étnicos cuyos polimorfismos genéticos sean diferentes a los incluidos en la muestra estudiada.
Una vez surtidos estos requisitos es frecuente encontrar grandes dificultades para la incorporación de la prueba en la práctica clínica y que ocupe el lugar que se merece para decidir a priori el medicamento o la dosis a prescribir. Varias pueden ser las razones por las cuales la implementación clínica de la información farmacogenómica ha sido mínima hasta ahora. Sin ignorar la resistencia del prescriptor para abandonar la estrategia de «ensayo y error» y su inseguridad para elegir fármacos o ajustar dosis con base en el perfil genético del paciente, es preciso admitir que una gran cantidad de medicamentos con margen de seguridad amplio tienen efectos fácilmente detectables mediante examen físico o paraclínicos sencillos y que, ante un evento indeseable o falla terapéutica, en muchos casos existe la opción de recurrir a medicamentos alternativos, sin mayores secuelas para el paciente; en tales circunstancias las pruebas farmacogenéticas son realmente inocuas. Por otro lado, muchos fármacos poseen vías de transporte o metabólicas paralelas y un transportador o una enzima funcional pueden compensar la proteína deficiente. Finalmente, una variante alélica con un impacto funcional comprobado puede no tener efectos clínicos manifiestos, o hacerlo sólo bajo ciertas circunstancias, porque factores medio ambientales, la comorbilidad del paciente y las interacciones farmacológicas pueden modificar el valor predictivo de muchos biomarcadores farmacogenómicos; no se debe perder de vista que las respuestas farmacológicas son fruto de una serie de variables genéticas y ambientales que interaccionan de manera compleja y que la información farmacogenómica puede ser sólo un elemento de juicio más para predecir una respuesta36.
De modo que la promesa de la farmacogenética de convertirse en un instrumento que contribuya a identificar «el fármaco adecuado, a las dosis adecuadas, en el individuo adecuado» únicamente se irá logrando a medida que se comprenda de qué manera las variaciones genómicas se traducen en variaciones en las respuestas a fármacos, y éstas, a su vez, tengan impacto clínico, en el sentido que tienen el potencial de mejorar la relación de seguridad/eficacia de un fármaco37. Los siguientes son algunos ejemplos donde la información farmacogenómica es potencialmente útil para la toma de decisiones terapéuticas:
Oncología. Tal vez en ningún otro campo de la medicina se ha necesitado con tanta urgencia el descubrimiento de nuevos agentes con mayor selectividad de acción y el desarrollo de protocolos de tratamiento con mayor poder predictivo como en la quimioterapia del cáncer. Los costos del tratamiento, el uso de fármacos citotóxicos con estrecho margen de seguridad, la gran variabilidad individual en la tolerabilidad de los pacientes y en la respuesta al tratamiento, las secuelas devastadoras de las fallas terapéuticas, etc, han hecho del manejo del cáncer un terreno propicio para beneficiarse del descubrimiento de marcadores farmacogenómicos38.
Los dos escenarios de la farmacogenómica, la genotipificación del paciente y la genotipificación del tumor, se perfilan como poderosos instrumentos que contribuirán a eliminar muchas de las incertidumbres y fracasos de la estrategia anticancerosa tradicional. En el primer caso, ya se han puesto de ejemplo las potenciales consecuencias clínicas del fenotipo metabolizador del paciente, en lo que respecta a tamoxifen y tioureas. Por otro lado, la caracterización farmacogenómica de muchos tipos de cáncer ha hecho posible el desarrollo de una nueva generación de agentes capaces de dirigirse en forma altamente selectiva contra moléculas críticas del proceso neoplásico y ha posibilitado la estratificación de los pacientes con base en la genética del tumor, mejorando significativamente la relación riesgo/beneficio del manejo de muchos tipos de cáncer; tal es el caso de trastuzumab, cetuximab y dasatinib, ya mencionados38.
Esta identificación de marcadores genéticos de susceptibilidad al tratamiento ha exigido incluso reformular la estrategia de investigación de nuevos medicamentos anticancerosos, pues con frecuencia no se puede extrapolar la respuesta de células tumorales a partir del genotipo hecho sobre células normales (casi siempre linfocitos) del paciente, debido a la presencia de aberraciones cromosómicas y aneuploidías (número variable de cromosomas) en células neoplásicas, en cuyo caso tales células podrían responder en forma diferente a como lo hacen células normales39.
Infección por VIH. El SIDA es un proceso crónico que requiere tratamiento de por vida, con complejas combinaciones de medicamentos potentes y de estrecho margen de seguridad. Es de esperar entonces que la farmacogenómica juegue aquí un papel importante, aunque muchos descubrimientos están todavía en fase de investigación y es necesario vencer numerosos obstáculos antes de que tengan aplicabilidad clínica. La variabilidad interindividual en los resultados del tratamiento antirretroviral es consecuencia de una multitud de factores del huésped y del virus, aunque el gran progreso en la farmacogenómica del VIH ha ocurrido en el estudio y aplicación de las bases genéticas de la susceptibilidad o resistencia del virus a los agentes antirretrovirales40, tópico que no será objeto de tratamiento en esta revisión.
Hasta ahora sólo unos pocos polimorfismos genéticos del paciente están claramente asociados con respuestas indeseables a los antirretrovirales. Estos son algunos ejemplos41-44:
1. Hipersensibilidad: El abacavir es un inhibidor nucleósido de la transcriptasa inversa, de amplio uso en el tratamiento de la enfermedad. Entre 3% y 6% de los usuarios puede provocar una reacción de hipersensibilidad potencialmente letal, que contraindica el uso subsiguiente del medicamento. Esta reacción se asocia con el alelo HLA-B*5701 del correspondiente gen del complejo mayor de histocompatibilidad. La genotipificación HLA-B*5701 es recomendada por las autoridades sanitarias como prueba previa a la prescripción por primera vez o al reinicio del fármaco. Como tamizaje pretratamiento en caucásicos e hispanos es costo-efectivo; en poblaciones orientales y negras, donde la prevalencia del alelo HLA-B*5701 es menor, aún no se ha definido la relevancia clínica del estudio.
2. Desorden de los lípidos: Se hallan comprometidos cinco genes que influyen en los niveles séricos de lípidos, con variantes de APOE y APOC3 como principales factores de riesgo de dislipidemia (sobre todo hipertrigliceridemia) asociada con antirretrovirales, en particular con el ritonavir. Se estima que si se implementara la estrategia de seleccionar el tratamiento antirretroviral de acuerdo con el genotipo podría reducirse en 30% el número de personas que desarrollan hipertrigliceridemia.
3. Desórdenes mitocondriales: Los trastornos metabólicos de la toxicidad mitocondrial provocada por agentes antirretrovirales, en particular los inhibidores de la transcriptasa inversa nucleótidos, se atribuyen a la inhibición de la transcripción de genes que codifican enzimas de la fosforilación oxidativa, debido a la acumulación de mutaciones en el ADN mitocondrial (mtDNA); aún no se ha descrito el genotipo de riesgo para esta toxicidad.
4. Hiperbilirrubinemia: Es un efecto adverso de indinavir (IDV) y atazanavir (ATV), asociado con alelos defectuosos del gen del transportador de la bilirrubina no conjugada al interior de la célula (gen SLCO1B1) y del gen de la UDP-glucuronosiltransferasa 1A1 (UGT1A1), la enzima encargada de conjugar la bilirrubina. Las frecuencias de estos alelos varían entre los grupos étnicos, razón por la cual la ictericia por IDV y ATV es más frecuente en negros y muy rara en orientales.
5. Neurotoxicidad: La efectividad y toxicidad del efavirenz depende críticamente de la actividad de la enzima CYP2B6, encargada de su degradación; a concentraciones séricas bajas (<1 mg/l) se asocia con resistencia y falla terapéutica, y a concentraciones altas (>4 mg/l) con riesgo incrementado de trastornos del SNC.
Inmunología de trasplantes. Algunos polimorfismos genéticos dividen la población en individuos con alta, media o baja capacidad de producción de mediadores de respuesta inmune, y ciertos fenotipos de «alta inmunidad» se asocian con predisposición al rechazo de órganos trasplantados. Además, los fármacos inmunosupresores tienen margen de seguridad estrecho, de modo que la subdosis se asocia con rechazo del órgano trasplantado y la sobredosis con inmunosupresión; en estos casos la farmacogenética podría contribuir a la selección del fármaco y las dosis sobre una base más individualizada, con mayor probabilidad de éxito. Los siguientes son algunos ejemplos de agentes inmunosupresores sobre los cuales hay evidencia de que ciertos polimorfismos genéticos influyen sobre los resultados del tratamiento45,46:
1. Ácido micofenólico: es un inhibidor suicida de la enzima inosin monofosfato deshidrogenasa (IMPDH), de mucha importancia en la proliferación y funciones de linfocitos B y T. Este agente es muy vulnerable al impacto de las variantes genéticas que codifican transportadores, enzimas y receptores involucrados en su absorción, transporte, metabolismo y mecanismo de acción; pocas veces un mismo fármaco está sujeto a tanta variedad de genes altamente polimórficos, con actividad aumentada, normal, deficiente o nula. Sin embargo, todavía no hay consenso acerca de la relevancia clínica de la farmacogenética del ácido micofenólico, y bien podría ser este un buen ejemplo de interacciones complejas de fármaco-genes, donde las consecuencias fenotípicas de algunos polimorfismos pudieran ser contrarrestadas por otros polimorfismos. Esta es una de las razones por las cuales los análisis haplotípicos (toma en cuenta el efecto combinado de varios alelos) aumentan la probabilidad de detectar biomarcadores farmacogenómicos potencialmente útiles en la práctica clínica.
2. Inhibidores de calcineurina: La ciclosporina y el tacrolimus impiden la activación de las células T. Aunque la relevancia clínica de la farmacogenética de estos agentes no ha sido establecida en forma concluyente, ya se conocen algunos hechos. Por ejemplo, el fenotipo alto productor de TGF-a1 (factor-a1 de crecimiento transformante) se asocia con incremento en el riesgo de nefrotoxicidad severa por ciclosporina en personas con trasplante cardíaco. También se halló fuerte asociación entre genotipo deficiente del transportador glicoproteína-P del donante y nefrotoxicidad por ciclosporina en el recipiente. En forma similar, algunos estudios han encontrado que en el trasplante de hígado tanto el genotipo metabolizador del recipiente como el del donante afectan la farmacocinética del tacrolimus; en efecto, los pacientes que reciben hígados de donantes con el genotipo CYP3A5 nativo (no mutado) requieren mayores dosis para alcanzar niveles sanguíneos terapéuticos de tacrolimus, que los pacientes recipientes de hígados de donantes con genotipo metabolizador deficiente, aunque el genotipo del recipiente influye más que el genotipo del donante.
3. Azatioprina: Actúa como antimetabolito e inhibe la síntesis de purinas. Como ya se dijo es degradada por la tiopurina metil-transferasa (TPMT), una enzima polimórfica que divide a la población en metabolizadores rápidos, intermedios y lentos, con consecuencias en términos de efectividad y seguridad: a dosis útiles y bien toleradas por los metabolizadores rápidos, los metabolizadores lentos tienen mayor riesgo de mielotoxicidad y de sobre-inmunosupresión. La FDA recomienda la gentotipificación TPMT en pacientes candidatos a tratamiento con azatioprina.
Varios. Pueden citarse muchos otros ejemplos donde la farmacogenómica ofrece una herramienta potencialmente útil en la toma de decisiones médicas, con fármacos que se caracterizan por tener eficacia y toxicidad impredecibles, por sus grandes variaciones individuales relacionadas con las dosis efectivas, o por poseer tiempos de latencia terapéutica prolongados. Este es el caso de los antiepilépticos, los antidepresivos, los antipsicóticos y los colinérgicos útiles en la enfermedad de Alzheimer (rivastigmina, donepecilo, etc)47,48, cuyas variaciones farmacogenéticas parecen ser importantes en la determinación de su eficacia, tolerabilidad y seguridad, aunque ha sido difícil precisar algunos casos. Igualmente es deseable la identificación prospectiva de los pacientes que tolerarán y responderán a fármacos como los medicamentos antifactor de necrosis tumoral-a (etanercept, infliximab, adalimumab), que son costosos, actúan tardíamente y sólo cerca de 60% de los pacientes responde a ellos49.
IMPACTO EN EL DESARROLLO DE FÁRMACOS
Históricamente, más de 90% de las moléculas estudiadas como fármacos potenciales nunca llegaron al mercado por eficacia insuficiente o toxicidad inaceptable en animales o humanos. Incluso, después de haber sido lanzados al mercado, algunos medicamentos debieron ser retirados por toxicidad inesperada, como ocurrió hace algunos años con astemizol, terfenadina, fenfluramina y mibefradil. Esto muestra la ineficiencia del modelo experimental convencional para el desarrollo de fármacos y fue un llamado de atención para la implementación de nuevas técnicas más eficientes y con mayor poder predictivo50. El cambio del «paradigma de la química» (se buscan deliberadamente o se encuentran de manera accidental moléculas con efectos medicinales y prácticamente lo último que se conoce de ellas es cómo actúan), al «paradigma de la biología» (se caracteriza molecularmente una ruta metabólica o una función y se interviene en un lugar crítico, sintetizando la molécula que module la ruta, bloquee o estimule la función3,7) ha mejorado el rendimiento y acortado los tiempos de investigación farmacológica.
La farmacogenómica no sólo tiene el potencial de influir en la eficacia y la seguridad, sino que se está convirtiendo en una fuente de desarrollo de medicamentos aún más rápida y eficiente. En efecto, para la industria farmacéutica la farmacogenómica brinda la oportunidad de descubrir mejores fármacos, detectar oportunamente a los pacientes con la predisposición genética a una buena respuesta y reducir significativamente costos del proceso experimental preclínico y clínico. Actualmente, en forma simultánea con los estudios de seguridad y eficacia, se llevan a cabo sub-estudios genómicos exploratorios que se proponen identificar biomarcadores predictores de respuestas farmacológicas de interés (terapéuticas o indeseables); estos estudios parten de la premisa de que el perfil de expresión génica de los pacientes que responden de una manera a un tratamiento, es diferente del perfil de expresión génica de quienes responden distinto.
Durante las fases tempranas de investigación el descubrimiento de un biomarcador que permita estratificar la población de enfermos en subgrupos de acuerdo a la respuesta terapéutica, hace posible excluir anticipadamente los pacientes que responderán de forma inadecuada al agente en estudio, con la consecuente reducción de fallas terapéuticas, reacciones adversas y costos de investigación. Así, fármacos que pudieran haber sido abandonados por problemas de seguridad/eficacia en el conjunto de enfermos, podrán ser aprobados para subgrupos de enfermos, beneficiando a pacientes, prescriptores, autoridades sanitarias y a la propia industria farmacéutica. Además, se reduce la probabilidad de retiros post-mercadeo debido a efectos indeseables, al permitir encontrar a tiempo los pacientes de alto riesgo51-53.
El término teragnóstico se refiere a pruebas diagnósticas ligadas directamente con una terapia específica, es decir, una estrategia de tratamiento individualizado en función de una prueba farmacogenómica que identifica el paciente que probablemente se beneficiará o perjudicará con el fármaco. Una vía rápida y una forma razonable de evitar un tratamiento inútil o inaceptablemente riesgoso, que le abre las puertas a un esquema de trabajo mancomunado entre la industria farmacéutica y la industria diagnóstica, para el co-desarrollo y la aprobación simultánea de nuevos fármacos con sus respectivos marcadores farmacogenómicos que predicen respuesta53.
CONSIDERACIONES ÉTICAS Y LEGALES
Los avances en el campo de la farmacogenómica han llevado a las autoridades sanitarias y oficiales a considerar su impacto a nivel social, ético y legal y a tomar acciones regulatorias. Para mayor información al respecto se recomienda consultar la página de la OMS http://who.int/genomics/elsi/en/
Aunque la farmacogenética es una disciplina ligada con menores problemas éticos que otras aplicaciones médicas de la genética, tales como el diagnóstico presintomático de enfermedades sin tratamiento actual, porque se enfoca en los polimorfismos relacionados con respuesta a fármacos, por lo general no asociados con enfermedad; desde luego toda prueba farmacogenómica debe contar con el consentimiento informado del paciente, y de todos modos los resultados de estas pruebas deben ser mantenidos en forma confidencial y los pacientes protegidos del riesgo de estigmatización o discriminación como resultado de la prueba; por la misma razón, la incorporación de minorías étnicas en algunos estudios farmacogenómicos debe ser objeto de consideraciones especiales1.
No obstante debe hacerse una distinción, porque las pruebas no asociadas con factores de riesgo o enfermedad (por ejemplo, el genotipo metabolizador) resultan menos inquietantes con respecto a la confidencialidad del paciente, riesgo de mal uso de la información y posibilidad de estigmatización, que aquellas pruebas predictoras de respuesta a tratamientos (ejemplo, el her-2 en cáncer de mama), pues la posterior prescripción o no del fármaco a subgrupos de pacientes, prácticamente dejará al descubierto su condición genética. También podría resultar estigmatizada la persona con baja probabilidad de responder a un tratamiento, aunque también tenga baja probabilidad de desarrollar la enfermedad, sobre todo si el estado de no respondedor se asocia con enfermedad crónica de alto costo3. El hecho de resultar inelegible para un tratamiento puede generar ansiedad y temores y podría tener un impacto negativo en asuntos de seguros y empleo54.
Desde el punto de vista investigativo, por la naturaleza sensible de la información genética, los estudios farmacogenómicos que generalmente acompañan a los ensayos clínicos farmacológicos deben recibir un trato especial, con consentimiento informado separado y declaración expresa de anonimización de muestras biológicas y datos, tiempo de conservación de las muestras y consejería genética, entre otros. El hecho de identificar los subgrupos de pacientes con predisposición genética a una buena respuesta terapéutica plantea el interrogante de ¿qué hacer con aquellos que portan un «genotipo probablemente no respondedor»?3
En lo que toca con el prescriptor, en la medida que se incremente el número de marcadores farmacogenómicos validados clínicamente, aumentará el número de fármacos con indicaciones, precauciones o contraindicaciones relacionadas con la presencia o ausencia de un biomarcador genético. Esta tendencia implica que los clínicos tendrán que enfrentarse a información farmacogenómica al momento de prescribir algunos medicamentos o ajustar unas dosis. De modo que en casos bien definidos no está lejos el día en que podrá ser considerado no ético exponer a un individuo a un fármaco (o a una dosis) inefectivo o peligroso, cuando la respuesta hubiera podido ser prevista con un prueba farmacogenética. Ansell SM et al.55 citan el caso de una persona que había muerto por arritmia cardíaca desencadenada por un síndrome de QT largo. La historia clínica y electrocardiográfica de una hermana asintomática de 18 años fue revisada por ocho cardiólogos especializados en electrofisiología, y sólo dos de ellos conceptuaron que debería ser tratada con beta-bloqueadores. Enseguida se les reveló a los médicos el genotipo de la joven, que mostraba su condición de portadora de la misma mutación causante de la muerte de su hermana. Todos los cardiólogos recomendaron terapia con beta-bloqueadores y tres de ellos sugirieron además la implantación de desfibrilador. Este ejemplo, que puede hacerse extensivo a muchas circunstancias médicas, muestra cómo una simple prueba genética es capaz de provocar cambios radicales en el abordaje terapéutico propuesto.
Como ya se ha mencionado, las grandes diferencias inter-étnicas en las prevalencias de los marcadores farmacogenómicos modifican la relación riesgo/beneficio de muchos medicamentos en función de los grupos étnicos, pues la relevancia clínica del efecto benéfico o indeseable dependerá de la frecuencia del marcador en cada población. Las implicaciones éticas y legales del cambio en la relación de seguridad/eficacia de los medicamentos de acuerdo con la impronta genética de cada grupo étnico también son claras y deberían ser tenidas en cuenta por prescriptores y autoridades sanitarias. Finalmente, esta variabilidad inter-étnica también se debe considerar por los organismos oficiales encargados de los programas de farmacovigilancia, como por las compañías farmacéuticas en sus planes de mercadeo17.
NUEVAS PERSPECTIVAS
Ninguna revisión de las relaciones entre genes y medicamentos está completa si no se mencionan, aunque sea en forma somera, dos novedosos mecanismos de regulación de la expresión génica, llamados a convertirse en una rica fuente de nuevos medicamentos y de interesantes descubrimientos en el campo de la farmacogenómica. En primer lugar, ahora sabemos que las exposiciones ambientales no solo pueden producir cambios en el genoma (mutaciones) y causar malformaciones y otras enfermedades, sino que pueden inducir cambios en la expresión génica sin modificar la secuencia del ADN. La epigenética tiende un nuevo puente que conecta el ambiente y el genoma al explicar la expresión de genes por mecanismos que no están codificados en el ADN. Estos cambios ADN-independientes, debidos a metilación de ADN, modificación de histonas en la cromatina y microRNAs (miRNAs), desempeñan un papel fundamental en los patrones de expresión génica durante el desarrollo y diferenciación normales, pero también juegan su papel en una variedad de respuestas biológicas y en el desarrollo de muchas enfermedades, notablemente del cáncer donde, por ejemplo, es común encontrar hipermetilación en la región del promotor de genes supresores de tumores, asociada con la pérdida de la capacidad de expresión del gen. De otra parte, muchos genes que codifican enzimas, transportadores, receptores, segundos y terceros mensajeros involucrados con el destino o la acción de fármacos están bajo control epigenético. La farmacoepigenómica es un nuevo campo de la ciencia que empieza a encontrar explicaciones a respuestas farmacológicas no explicadas por la farmacogenómica53,56-58.
El segundo mecanismo tiene que ver con un grupo de receptores nucleares que empiezan a ser reconocidos como reguladores de la transcripción, porque se unen a sitios específicos del genoma y controlan la expresión de una gran cantidad de genes. Entre los reguladores de este tipo mejor caracterizados se encuentran el «pregnane X receptor» (PXP), los «retinoid-related orphan receptors» (RORs), el «constitutive androstane receptor» (CAR), los «liver X receptors» (LXRs), los «peroxisome proliferator-activated receptors» (PPARs) y el «vitamin D receptor» (VDR). La expresión de genes que codifican enzimas y transportadores de fármacos es coordinada por una compleja red de estos factores, que actúan como sensores de xenobióticos potencialmente tóxicos y envían señales para su eliminación. Aunque todavía no están suficientemente estudiados, se acepta que los polimorfismos genéticos de estos receptores nucleares pueden influir en su actividad reguladora, en su potencial de inducir o inhibir enzimas y transportadores y en la severidad de ciertas interacciones farmacológicas40,59.
REFERENCIAS
1. Brockmoller J, Tzvetkov MV. Pharmacogenetics: data, concepts and tools to improve drug discovery and drug treatment. Eur J Clin Pharmacol. 2008; 64: 133-57. [ Links ]
2. Kalow W. Pharmacogenetics in biological perpective. Pharmacological Rev. 1997; 49: 369-80. [ Links ]
3. Lindpaintner K. The impact of pharmacogenetics and pharmacogenomics on drug discovery. Nat Rev Drug Discov. 2002; 1: 463-9. [ Links ]
4. Palmer LJ, Cardon LR. Shaking the tree: mapping complex disease genes with linkage disequilibrium. Lancet. 2005; 366: 1223-34. [ Links ]
5. Peters EJ, McLeod HL. Ability of whole-genome SNP arrays to capture 'must have' pharmacogenomic variants. Pharmacogenomics. 2008; 9: 1573-7. [ Links ]
6. Altman RB, Klein TE. Challenges for biomedical informatics and pharmacogenomics. Annu Rev Pharmacol Toxicol. 2002; 42: 113-33. [ Links ]
7. Relling MV, Giacomini KM. Pharmacogenetics. In: Brunton LL, Lazo JS, Parker KL, (eds). Goodman and Gilman's. The Pharmacological Basis of Therapeutics. 11ª ed. McGraw-Hill; 2006. p. 93-115. [ Links ]
8. Bhathena A, Spear BB. Pharmacogenetics: improving drug and dose selection. Curr Opin Pharmacol. 2008; 8: 639-46. [ Links ]
9. Dorn GW2nd, Cresci S. The mechanistic imperative for pharmacogenomics. Pharmacogenomics. 2008; 9: 801-3. [ Links ]
10. Johnson JA. Ethnic differences in cardiovascular drug response: potential contribution of pharmacogenetics. Circulation. 2008; 118: 1383-93. [ Links ]
11. González FJ, Nebert DW. Evolution of the P450 gene superfamily: animal-plant «warfare», molecular drive and human genetic differences in drug oxidation. Trends Genet. 1990; 6: 182-6. [ Links ]
12. González FJ, Tukey RH. Drug metabolism. In: Brunton LL, Lazo JS, Parker KL, (eds). Goodman and Gilman's. The Pharmacological Basis of Therapeutics. 11ª ed. McGraw-Hill; 2006. p. 71-91. [ Links ]
13. Whitlock JP. Induction of cytochrome P4501A1. Annu Rev Pharm Toxicol. 1999; 39: 103-25. [ Links ]
14. Michalelets EL. Update: Clinically significant cytochrome P-450 drug interactions. Pharmacotherapy. 1998; 18: 84-112. [ Links ]
15. Daly AK, Brockmoller J, Broly F, Eichelbaum M, Evans WE, González FJ, et al. Nomenclature for human CYP2D6 alleles. Pharmacogenetics. 1996; 6: 193-201. [ Links ]
16. Isaza MC, Isaza MG, Fuentes GJ, Marulanda MT. Fundamentos de farmacología en terapéutica. 5ª ed. Pereira: Postergraph; 2008. [ Links ]
17. Zanger UM, Turpeinen M, Klein K, Schwab M. Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal Bioanal Chem. 2008; 392: 1093-108. [ Links ]
18. Isaza C, Henao J, Isaza JH, Sepúlveda JC, Beltrán L. Phenotype-genotype analysis of CYP2C19 in Colombian mestizo individuals. BMC Clin Pharmacol. 2007; 7: 6. [ Links ]
19. Klotz U. Clinical impact of CYP2C19 polymorphism on the action of proton pump inhibitors: a review of a special problem. Int J Clin Pharmacol Ther. 2006; 44: 297-302. [ Links ]
20. Isaza CA, Henao J, López AM, Cacabelos R. Isolation, sequence and genotyping of the drug metabolizer CYP2D6 gene in the Colombian population. Methods Find Exp Clin Pharmacol. 2000; 22: 695-705. [ Links ]
21. Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J. 2005; 5: 6-13. [ Links ]
22. Chou WH, Yan FX, de Leon J, Barnhill J, Rogers T, Cronin M, et al. Extension of a pilot study: impact from the cytochrome P450 2D6 polymorphism on outcome and costs associated with severe mental illness. J Clin Psychopharmacol. 2000; 20: 246-51. [ Links ]
23. Meyer UA, Zanger UM. Molecular mechanisms of genetic polymorphisms of drug metabolism. Annu Rev Pharmacol Toxicol. 1997; 37: 269-96. [ Links ]
24. Isaza C, Henao J, López AM, Cacabelos R. Allelic variants of the thiopurine methyltransferase (TPMT) gene in the Colombian population. Methods Find Exp Clin Pharmacol. 2003; 25: 423-9. [ Links ]
25. van den Akker-van Marle ME, Gurwitz D, Detmar SB, Enzing CM, Hopkins MM, Gutiérrez de Mesa E, et al. Cost-effectiveness of pharmacogenomics in clinical practice: a case study of thiopurine methyltransferase genotyping in acute lymphoblastic leukemia in Europe. Pharmacogenomics. 2006; 7: 783-92. [ Links ]
26. Xie HG, Kim RB, Wood AJ, Stein CM. Molecular basis of ethnic differences in drug disposition and response. Annu Rev Pharmacol Toxicol. 2001; 41: 815-50. [ Links ]
27. Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007; 117: 1422-31. [ Links ]
28. Wang L, Weinshilboum RM. Pharmacogenomics: candidate gene identification, functional validation and mechanisms. Hum Mol Genet. 2008; 17(R2): R174-9. [ Links ]
29. Becquemont L. Evidence for a pharmacogenetic adapted dose of oral anticoagulant in routine medical practice. Eur J Clin Pharmacol. 2008; 64: 953-60. [ Links ]
30. International Warfarin Pharmacogenetics Consortium, Klein TE, Altman RB, Eriksson N, Gage BF, Kimmel SE, Lee MT, et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N Engl J Med. 2009; 360: 753-64. [ Links ]
31. Isaza C, Henao J, Ramírez E, Cuesta F, Cacabelos R. Polymorphic variants of the beta2-adrenergic receptor (ADRB2) gene and ADRB2-related propanolol-induced dyslipidemia in the Colombian population. Methods Find Exp Clin Pharmacol. 2005; 27: 237-44. [ Links ]
32. Isaza CA, Henao J, Sánchez JC, Porras GL, Cardona J, Bedoya G. Beta-2-adrenergic receptor polymorphisms and changes in lipids induced by metoprolol. Pharmacology. 2007; 80: 279-85. [ Links ]
33. Nguyen TV, Center JR, Eisman JA. Pharmacogenetics of osteoporosis and the prospect of individualized prognosis and individualized therapy. Curr Opin Endocrinol Diabetes Obes. 2008; 15: 481-8. [ Links ]
34. Woodcock J, Lesko LJ. Pharmacogenetics -tailoring treatment for the outliers. N Engl J Med. 2009; 360: 811-3. [ Links ]
35. Marsh S. Pharmacogenetics: global clinical markers. Pharmacogenomics. 2008; 9: 371-3. [ Links ]
36. Dempfle A, Scherag A, Hein R, Beckmann L, Chang-Claude J, Schäfer H. Gene-environment interactions for complex traits: definitions, methodological requirements and challenges. Eur J Hum Genet. 2008; 16: 1164-72. [ Links ]
37. Zhou SF, Di YM, Chan E, Du YM, Chow VD, Xue CC, et al. Clinical pharmacogenetics and potential application in personalized medicine. Curr Drug Metab. 2008; 9: 738-84. [ Links ]
38. Nishiyama M, Eguchi H. Recent advances in cancer chemotherapy: current strategies, pharmacokinetics, and pharmacogenomics. Adv Drug Deliv Rev. 2008; 61: 367-68. [ Links ]
39. Cheng Q, Yang W, Raimondi SC, Pui CH, Relling MV, Evans WE. Karyotypic abnormalities create discordance of germline genotype and cancer cell phenotypes. Nat Genet. 2005; 37: 878-82. [ Links ]
40. Telenti A, Zanger UM. Pharmacogenetics of anti-HIV drugs. Annu Rev Pharmacol Toxicol. 2008; 48: 227-56. [ Links ]
41. Tozzi V, Libertone R, Liuzzi G. HIV pharmacogenetics in clinical practice: recent achievements and future challenges. Curr HIV Res. 2008; 6: 544-54. [ Links ]
42. Mahungu TW, Johnson MA, Owen A, Back DJ. The impact of pharmacogenetics on HIV therapy. Int J STD AIDS. 2009; 20: 145-51. [ Links ]
43. Tozzi V, Libertone R, Liuzzi G. HIV pharmacogenetics in clinical practice: recent achievements and future challenges. Curr HIV Res. 2008; 6: 544-54. [ Links ]
44. Roca B. Pharmacogenomics of antiretrovirals. Med Clin (Barc). 2009; 132: 268-71. [ Links ]
45. de Jonge H, Kuypers DR. Pharmacogenetics in solid organ transplantation: current status and future directions. Transplant Rev. 2008; 22: 6-20. [ Links ]
46. Girnita DM, Burckart G, Zeevi A. Effect of cytokine and pharmacogenomic genetic polymorphisms in transplantation. Curr Opin Immunol. 2008; 20: 614-25. [ Links ]
47. Laqscher W, Klotz U, Zimprich F, Schmidt D. The clinical impact of pharmacogenetics on the treatment of epilepsy. Epilepsia. 2009; 50: 1-23. [ Links ]
48. Cacabelos R. Pharmacogenomics in Alzheimer's disease. Methods Mol Biol. 2008; 448: 213-357. [ Links ]
49. Ranganathan P. An update on pharmacogenomics in rheumatoid arthritis with a focus on TNF-blocking agents. Curr Opin Mol Ther. 2008; 10: 562-7. [ Links ]
50. Ryan TP, Stevens JL, Thomas CE. Strategic applications of toxicogenomics in early drug discovery. Curr Opin Pharmacol. 2008; 8: 654-60. [ Links ]
51. Malik NN. Drug discovery: past, present and future. Drug Discov Today. 2008; 13: 909-12. [ Links ]
52. McCarthy AD, Kennedy JL, Middleton LT. Pharmacogenetics in drug development. Philos Trans R Soc Lond B Biol Sci. 2005; 360: 1579-88. [ Links ]
53. Pene F, Courtine E, Cariou A, Mira JP. Toward theragnostics. Crit Care Med. 2009; 37 (1 Suppl): S50-8. [ Links ]
54. Ozdemir V, Graham JE, Godard B. Race as a variable in pharmacogenomics science: from empirical ethics to publication standards. Pharmacogenet Genomics. 2008; 18: 837-41. [ Links ]
55. Ansell SM, Ackerman MJ, Black JL, Roberts LR, Tefferi A. Primer on medical genomics. Part VI: Genomics and molecular genetics in clinical practice. Mayo Clin Proc. 2003; 78: 307-17. [ Links ]
56. Reamon-Buettner SM, Mutschler V, Borlak J. The next innovation cycle in toxicogenomics: environmental epigenetics. Mutat Res. 2008; 659: 158-65. [ Links ]
57. Gomez A, Ingelman-Sundberg M. Pharmacoepigenetics: its role in interindividual differences in drug response. Clin Pharmacol Ther. 2009; 85: 426-30. [ Links ]
58. Peedicayil J. Pharmacoepigenetics and pharmacoepigenomics. Pharmacogenomics. 2008; 9: 1785-6. [ Links ]
59. Wada T, Kang HS, Jetten AM, Xie W. The emerging role of nuclear receptor RORalpha and its crosstalk with LXR in xeno- and endobiotic gene regulation. Exp Biol Med. 2008; 233: 1191-201. [ Links ]

