










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombia Médica
On-line version ISSN 1657-9534
Colomb. Med. vol.41 no.3 Cali July/Sept. 2010
Evidence in Colombia of 625G>A polymorphism in the short chain acyl-CoA dehydrogenase gene, a variation which could cause glutaric aciduria in our populations
José Henry Osorio, PhD*
* Department of Basic Sciences for Health, Research Laboratory in Clinical Biochemistry and Molecular Pathology, Universidad de Caldas, Manizales, Colombia. e-mail: jose.osorio_o@ucaldas.edu.co
Received for publication June 12, 2009 Accepted for publication March 2, 2010
SUMMARY
Introduction: Short-chain acyl-CoA dehydrogenase (SCAD) is a homotetrameric mitochondrial flavoenzyme that catalyzes the initial reaction in short-chain fatty acid b-oxidation. The SCAD gene is located on chromosome 12q22 and is approximately 13 kb long with 10 exons and 1236 nucleotides of coding sequence. Hereditary SCAD deficiency has been reported and only a few cases of this disorder have been described.
Objective: The present study was conducted to determine the possible presence of the 625G>A variation in the short-chain acyl-CoA dehydrogenase gene in Caldas (Colombia), given that variations 625G>A and 511C>T are present in 14% of some studied populations; thereby sometimes causing its deficiency.
Methods: This is a descriptive study; blood samples from three-hundred adult volunteers were tested for 625G>A polymorphism, analysing the polymerase chain reaction amplified cDNA, using a single-stranded conformation polymorphism assay. The results were confirmed by direct bidirectional cycle sequencing using DNA from the positive persons.
Results: The polymorphism was identified and confirmed in four healthy persons.
Conclusion: This is evidence of the presence of 625G>A polymorphism in the short-chain acyl-CoA dehydrogenase gene in Colombia, meaning that some people in our populations can be at risk of suffering SCAD deficiency and its main complication: the ethylmalonic aciduria.
Keywords: Short-chain acyl-CoA dehydrogenase; 625G>A polymorphism; Ethylmalonic aciduria; b-oxidation.
Evidencia del polimorfismo 625G>A en el gen de la acil-CoA deshidrogenasa de cadena corta en Colombia, una variación que podría causar aciduria glutárica en algunas poblaciones del país
RESUMEN
Introducción: La acil-CoA deshidrogenasa de cadena corta (SCAD) es una flavoenzima homotetramérica mitocondrial que cataliza la reacción inicial de la â-oxidación de los ácidos grasos de cadena corta. El gen SCAD se ubica en el cromosoma 12q22, con una longitud de 13 kb, con 10 exones y 1236 nucleótidos de secuencia codificadora. Se ha informado la deficiencia hereditaria de SCAD y se han descrito pocos casos de la deficiencia.
Objetivo: El presente estudio buscó determinar la posible presencia del polimorfismo 625G>A en Caldas, Colombia, debido a que las variantes 625G>A y 511C>T en el gen de la acil-CoA deshidrogenasa de cadena corta están presentes en 14% de algunas poblaciones estudiadas, causando algunas veces su deficiencia.
Métodos: El presente estudio es descriptivo; se estudiaron muestras de sangre de 300 voluntarios para el polimorfismo 625G>A mediante la técnica de polimorfismo de conformación de la cadena simple, con ADN amplificado por reacción en cadena de la polimerasa. Los resultados se confirmaron por secuenciación.
Resultados: El polimorfismo se identificó en cuatro personas aparentemente sanas.
Conclusión: Existe evidencia de la presencia del polimorfismo 625 G>A en el gen de la acil-CoA en Colombia, lo que significa que algunas personas en las poblaciones del país pueden estar en riesgo de sufrir deficiencia de SCAD y su principal complicación, la aciduria etilmalónica.
Palabras claves: Acil-CoA deshidrogenasa de cadena corta; Polimorfismo 625G>A; Aciduria etilmalónica; β-oxidación.
All the currently identified mitochondrial fatty acid (FA) b-oxidation defects are autosomal recessive. The clinical manifestations result from the inability of FA-oxidising tissues to keep up with increased energy demands; therefore, target organs of FA oxidation defects include skeletal and cardiac muscles in addition to the liver1. Hypoketotic hypoglycaemia is present in nearly all the defects, and usually occurs following an intercurrent illness, but may occasionally be seen after a short fast. In general, FA oxidation disorders should always be included in the differential diagnosis of unexplained hypoglycaemia, metabolic acidosis, Reyes like syndrome, myopathy, recurrent myoglobinuria, and cardiomyopathy2. Laboratory findings reinforce the clinical diagnosis, levels of intermediary metabolites in urine (glucose, ketone bodies, lactate, pyruvate), and blood (non-sterified FA)3; urinary organic acid profile4; body fluids and tissue acylcarnitine analysis5; enzyme measurement and pathway intermediates in cultured cells and leukocytes6,7, and DNA analysis8 are used to confirm the diagnosis of any alteration.
Short-chain acyl-CoA dehydrogenase deficiency is a poorly characterized mitochondrial fatty acid b-oxidation disorder with a very variable clinical picture and at least 35 inactivating mutations and some polymorphic variants have been reported in the SCAD gene9. Hereditary SCAD deficiency was first reported in 198410 and only few cases of this disorder have been described for either classic SCAD deficiency or variant SCAD10-13. A clinically reliable diagnosis requires either a muscle biopsy for the measurement of the enzymatic SCAD activity or molecular genetic analysis of the SCAD gene, both only available in research laboratories. This situation is further complicated by the presence of the mainly two variants (625 G>A and 511C>T) in the SCAD gene that are frequent in the European population, and have been reported to confer disease susceptibility14. In the 625G>A variation the polymorphic site is a transition from G to A at position 625 (A625) of the coding region of the cDNA, changing a glycine to serine at amino acid position (G185S) of the precursor protein. The variant 625G>A has been shown to be associated with ethymalonic aciduria and other biochemical findings15. Because urinary ethymalonic acid (EMA) elevation most likely reflects a cellular accumulation of butyryl-CoA16, which is secondary to reduced SCAD catalytic activity, these patients are correctly considered as possibly having SCAD deficiency.
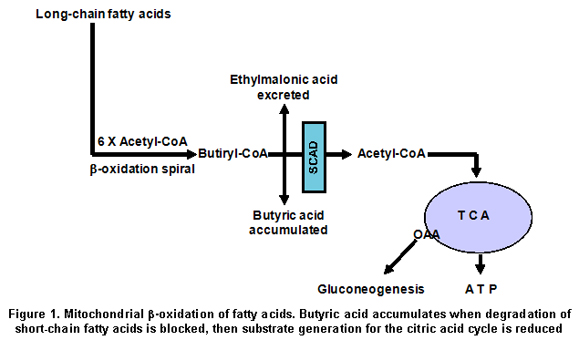
Despite the fact that most patients with EMA aciduria have not had their SCAD activity determined, an association between elevated EMA and SCAD has been documented by the presence of either or both of two SCAD gene susceptibility variations in 69% of patients with EMA aciduria17. The 625G>A variant shows homozygous prevalence of 60%, as SCAD protein presents reduced stability compared to the control SCAD protein18. As SCAD is a key enzyme in the oxidation of fatty acids, which serve as substrates for the gluconeogenesis (Figure 1), reduced SCAD activity because of the 625G>A variant for fasting hypoglycaemia, and ethymalonic aciduria as shown in the SCAD deficiency. The present study was conducted to determine the possible presence of the 625G>A variation in the short-chain acyl-CoA dehydrogenase gene in Caldas, Colombia, as the 625G>A and 511C>T variations are present in 14% of some populations studied, sometimes causing its deficiency.
MATERIALS AND METHODS
The present study is descriptive. Blood samples used in this study were from 300 adult, healthy volunteers between 18 and 49 years of age (120 men and 180 women), all born in Caldas, Colombia, apparently none suffering from any inherited inborn error of metabolism. According to the number of persons visiting our laboratory, samples were collected between January 2006 and January 2009, which means that samples were obtained by convenience, and all participants signed written consent.
Blood samples were extracted in tubes containing EDTA. DNA extraction was performed according to Gustafson et al.19, with some modifications. The polymorphism was identified by comparing the polymerase chain reaction (PCR) and amplified cDNA from a previously studied person carrying the polymorphism with the apparently healthy volunteers. A single-stranded conformation polymorphism (SSCP) assay, based on the following primers was used: Sense primer: 5-GCAGCTCTGAGAAAACCAC. Antisense primer: 5-ATGTCC AGGGTTTGCTGT. PCR Conditions: 3 min at 94°C, 40 sec at 94°C, 30 sec at 55°C, and 2 min at 72°C for 35 cycles with 500 ng of purified genomic DNA as template and 75 ng of each primer. When DNA fragments were subjected to electrophoresis in 8% acrilamide/bisacrilamide (19:1), 7. 5 M urea gel, at room temperature for 3 hours, the single-base change at position 625 was clearly detected after silver stain. The results were confirmed by direct bidirectional cycle sequencing using DNA.
According to Article 11 on its literal a from Resolution N° 8430 promulgated by the Ministry of Health for Scientific, technical and administrative guidelines for health research, the present study is considered without risk. The study was approved by the corresponding ethics committee.
RESULTS
After analyzing DNA samples from the 300 participants, the polymorphism was identified in four apparently healthy adult volunteers -all in heterozygosis. The first case was a 21-year-old male who was born from non-consanguineous parents, another two individuals were identified as carriers of the polymorphism; they are the parents of a family who presented two cases of sudden infant death after two consecutive pregnancies and they are cousins. After a third pregnancy, a baby was born and after 2 years she is still alive without problems; she did not present the 625G>A polymorphism. A fourth person was identified as carrying the polymorphism, an apparently normal 22-year-old woman. The four persons are from families without antecedents of any disease that could be related to some fatty acid oxidation disorder; the acylcarnitine analysis was normal for all of them.
DISCUSSION
Colombia is divided into 35 regions with an approximate total population of 45-million inhabitants, presenting racial mixture. Data obtained from the 2005 census shows that Caldas (a Colombian region) has a population of 968,740 inhabitants distributed within 29 municipalities20. For us, it is very important to detect the presence of this polymorphism to demonstrate the presence of fatty-acid-oxidation inherited diseases among us, and it is a valuable contribution for the future understanding of geographical distribution and ethnic origin elucidation of SCAD deficiency, as no previous reports have been published showing the presence of this polymorphism in these countries, while being mainly reported in Europe and the United States21. The first person presenting this variant in our study has a very interesting genetic charge for these kinds of works, as his father is from Tumaco, a region of black population and his mother is from the north of Antioquia, a place characterized by the presence of a strong Spaniard white genotype. The other 3 persons are from the centre of the country.
There are reports of ethnic differences observed with respect to the 625G>A variant, while the allele frequency was similar between Caucasians and Hispanics (25 and 30%, respectively), African-Americans and Asians carried the 625G>A variant less frequently (9 and 13%, respectively)22. Corydon et al.14, found the 625A variant allele in homozygous form in 60% of 135 patients with elevated EMA excretion, analysed because a suspicion of a metabolic disorder, compared with 7% occurrence in the general population. That is why, since about 10-14% of the general population is homozygous 625G>A or 511C>T or compound heterozygous for both, it is necessary to have some other indication that they are disease-associated. It is generally accepted that some biochemical analysis can be performed to confirm if the person carrying the polymorphism is suffering the disease. Because these patients are phenotypically diverse, the analysis of blood acylcarnitines is a good tool for diagnosing SCAD deficiency. The acylcarnitine profile in these patients is characterized by high blood concentrations of butyrylcarnitine (C4-acylcarnitine). The acylcarnitine profile for our patients was normal; hence, they are carrying the polymorphism without suffering the disease.
CONCLUSION
Short-chain acyl-CoA dehydrogenase (SCAD) deficiency is a clinically heterogeneous disorder. The clinical phenotype varies from fatal metabolic decompensation in early life to subtle adult onset, and some patients remain asymptomatic. Two mutations (511C>T; 625G>A) have been described in exons 5 and 6 of the SCAD gene, respectively. These variants are not true disease-causing mutations but can confer disease susceptibility because they alter the structural and catalytic properties of the SCAD protein. Although the prevalent finding for SCAD deficiency is neurological abnormalities, such as hypotonia and seizures accompanying general developmental delay, the population of patients with ethylmalonic aciduria with a high frequency of the 625A variant allele in the homozygous form also showed diverse phenotypes, typically neuromuscular symptoms, and hypoglycaemia. The acylcarnitine analysis is an important tool in diagnosing the disease, given that a high concentration of C4-acylcarnitine is consistent with a biochemical diagnosis of SCAD deficiency. This kind of study is important for us because it shows the presence of mutations or polymorphisms, which can cause some metabolic alterations related to enzymatic dysfunction of some biochemical ways, not reported or treated among us. We need to continue studying our populations to know the real frequency of this polymorphism because the principal studies have been performed mainly in Caucasoid populations.
Conflict of interest. None of the author had conflicts of interest related to this study.
REFERENCES
1. Stanley A. Carnitine disorders. Adv Pediatr. 1995; 42: 209-42. [ Links ]
2. Lteif AN, Schwenk WF. Hypoglycemia in infants and children. Endocr Metab Clin North Am. 1999; 23: 619-48. [ Links ]
3. Moser HW, Moser AB. Measurement of saturated very long chain fatty acids in plasma. In: Hommes FA (ed.). Techniques in diagnostic human biochemical genetics: a laboratory manual. New York: Wiley-Liss; 1991. p.177-92. [ Links ]
4. Greter J, Jacobson CE. Urinary organic acids: isolation and quantification for routine metabolic screening. Clin Chem. 1987; 33: 473-80. [ Links ]
5. Millington DS, Kodo N, Norwood DL, Roe CR. Tandem Mass Spectrometry: A new method for acylcarnitine profiling with potential for neonatal screening for inborn errors of metabolism. J Inher Metab Dis. 1990; 13: 321-4. [ Links ]
6. Pourfarzam M, Schaefer J, Turnbull DM, Bartlett K. Analysis of fatty acid oxidation intermediates in cultured fibroblasts to detect Mitochondrial oxidation disorders. Clin Chem. 1994; 40: 2267-75. [ Links ]
7. Schaefer J, Pourfarzam M, Bartlett K, Jackson S, Turnbull DM. Fatty acid oxidation in peripheral blood cells: characterisation and use for the diagnosis of fatty acid oxidation. Pediatr Res. 1995; 37: 345-60. [ Links ]
8. Ziadeh R, Hoffman EP, Finegold DN. Medium chain Acyl-CoA dehydrogenase deficiency in Pennsylvania: neonatal screening shows high incidence and unexpected mutation frequencies. Pediatr Res. 1995; 37: 675-8. [ Links ]
9. Jethva R, Bennett MJ, Vockley J. Short-chain acyl-coenzyme A dehydrogenase deficiency. Mol Gen Metab. 2008; 95: 195-200. [ Links ]
10. Turnbull DM, Bartlett K, Stevens DL, Alberti KGMM, Gibson GJ, Johnson MA, et al. Short-chain acyl-CoA dehydrogenase deficiency associated with a lipid-storage myophaty and secondary carnitine deficiency. N Engl J Med. 1984; 311: 1232-36. [ Links ]
11. Amendt BA, Greene C, Sweetman L, Cloherty J, Shih V, Moon A, et al. Short-chain acyl-coenzyme A dehydrogenase deficiency. Clinical and biochemical studies in two patients. J Clin Invest. 1987; 79: 1303-9. [ Links ]
12. Coates PM, Hale DE, Finocchiaro G, Tanaka K, Winter SC. Genetic deficiency of short-chain acyl-conzyme A dehydrogenase in cultured fibroblasts from a patient with muscle carnitine deficiency and severe skeletal muscle weakness. J Clin Invest. 1988; 81: 171-5. [ Links ]
13. Baerlocher KE, Steinmann B, Aguzzi A, Krähenbühl S, Roe CR, Vianey-Saban C. Short-chain acyl-CoA dehydrogenase deficiency in a 16-year-old girl with severe muscle wasting and scoliosis. J Inherit Metab Dis. 1997; 20: 427-31. [ Links ]
14. Corydon MJ, Gregersen N, Lehnert W, Ribes A, Rinaldo P, Kmoch S. Ethylmalonic aciduria is associated with an amino acid variant of short-chain acyl-coenzyme A dehydrogenase. Pediatr Res. 1996; 39: 1059-66. [ Links ]
15. Burlina AB, Dionisi-Vici C, Bennett MJ, Gibson KM, Servidei S, Bertini E, et al. A new syndrome with ethylmalonic aciduria and normal fatty acid oxidation in fibroblasts. J Pediatr. 1994; 124: 79-86. [ Links ]
16. Hegre CS, Halenz DR, Lane MD. The enzymatic carboxilation of butyryl-coenzyme A. J Am Chem Soc. 1959; 81: 6526-7. [ Links ]
17. Moczulski D, Majak I, Mamczur D. An overview of beta-oxidation disorders. Postepy Hig Med Dosw. (Online) 2009; 8: 266-77. [ Links ]
18. van Maldegem BT, Durán M, Wanders RJ, Niezen-Koning KE, Hogeveen M, Ijlst L, et al. Short-chain acyl-CoA dehydrogenase deficiency (SCADD): relatively high prevalence in the Netherlands and strongly variable fenotype; neonatal screening not indicated. Ned Tijdschr Geneeskd. 2008; 26: 1678-85. [ Links ]
19. Gustafson S, Proper JA, Bowie EJW, Sommer SS. Parameters affecting the yield of DNA from human blood. Biochemistry. 1987; 165: 294-9. [ Links ]
20. Departamento Nacional de Estadística (DANE). Colombia Censo General 2005. [fecha de acceso: enero 20 de 2009]. URL disponible en: http://www.dane.gov.co/censo/ [ Links ]
21. Gregersen N, Andresen BS, Corydon MJ, Corydon TJ, Olsen RK, Bolund L, et al. Mutation analysis in mitochondrial fatty acid oxidation defects: Exemplified by acyl-CoA dehydrogenase deficiencies, with special focus on genotype-phenotype relationship. Hum Mutat. 2001; 18: 169-89. [ Links ]
22. Corydon MJ, Vockley J, Rinaldo P, Rhead WJ, Kjeldsen M, Winter V, et al. Role of common gene variations in the molecular pathogenesis of short-chain acyl-CoA dehydrogenase deficiency. Pediatr Res. 2001; 49: 18-23. [ Links ]

