










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombia Médica
On-line version ISSN 1657-9534
Colomb. Med. vol.41 no.4 Cali Oct./Dec. 2010
Viral recognition by the innate immune system: the role of pattern recognition receptors*
Silvia Torres Pedraza, Microbiol1,2, Juan Guillermo Betancur, Biol2,3, Silvio Urcuqui-Inchima, Biol, PhD2
* This work was funded by Colciencias grant code number 111540820517-2.
1. PhD student, Corporación Ciencias Básicas Biomédicas, Universidad de Antioquia. e-mail: mayitator@gmail.com
2. Immunovirology Group, Sede de Investigación Universitaria, Universidad de Antioquia, Medellín, Colombia.
e-mail: silviourcuqui@gmail.com
3. PhD student, Institute of Molecular and Cellular Biosciences, The University of Tokyo, Japan.
e-mail: betancur@iam.u-tokyo.ac.jp
SUMMARY
Pattern recognition receptors are the main sensors of the innate immune response. Their function is to recognize pathogen-associated molecular patterns, which are molecules essential for the survival of microbial pathogens, but are not produced by the host. The recognition of pathogen-associated molecular patterns by pattern recognition receptors leads to the expression of cytokines, chemokines, and co-stimulatory molecules that eliminate pathogens, such as viruses, for the activation of antigen presenting cells and for the activation of specific adaptive immunity. Among the most thoroughly studied pattern recognition receptors implicated in viral infections, there are the toll-like receptors (TLRs) and the RNA helicase-type retinoic acid-inducible gene-1 receptors [or RIG-like receptors (RLRs)]. Moreover, other proteins such as PKR, 2-5 OAS, and ADAR also act as effector proteins in antiviral responses. The identification and characterization of pattern recognition receptors have contributed to our knowledge of the role of innate immunity in viral infections and has led us to better understand host-pathogen interactions. The most recent findings concerning the role of TLRs and RLRs in viral infections, the molecular mechanisms of viral ligand recognition through pattern recognition receptors, and the activation of their signaling pathways are discussed in this review.
Keywords: Innate immunity; Pro-inflammatory cytokines; Toll-like receptors; RIG-I-like receptors.
Reconocimiento viral por el sistema inmune innato: papel de los receptores de reconocimiento de patrones
RESUMEN
Los receptores de reconocimiento de patrones (PRR) son los principales sensores de la respuesta inmune innata. Su función es reconocer moléculas indispensables para la sobrevivencia de los patógenos, conocidas como patrones moleculares asociados a patógenos (PAMP). El reconocimiento de los PAMP por los PRR conlleva a la expresión de citoquinas, quimioquinas, y moléculas coestimuladoras implicadas en la eliminación de patógenos como virus, en la activación de células presentadoras de antígenos y en la inducción de una inmunidad adaptativa específica. Entre los PRR mejor descritos y con implicaciones en infecciones virales se encuentran los receptores tipo toll (TLR) y receptores tipo RNA helicasas inducibles por ácido retinoico (RLR); además las proteínas efectoras PKR, 2´-5´ OAS y ADAR también participan activamente en la respuesta antiviral. La descripción y caracterización de los PRR ha contribuido enormemente al entendimiento del papel de la respuesta inmune innata en las infecciones virales y han sido usados para comprender mejor las interacciones hospedero-patógenos. Se discuten en la presente revisión los más recientes conocimientos de los TLR y RLR, el mecanismo de reconocimiento de los virus vía PRRs y las vías de señalización activadas por dicho reconocimiento.
Palabras clave: Inmunidad innata; Citoquinas proinflamatorias; Receptores tipo toll; Receptores inducibles por ácido retinoico.
Innate immunity is the first protecting barrier of the host against foreign agents; its response and quality determine the development of adaptive immunity responses. The activation of the innate immune response involves a wide spectrum of cells and soluble factors that recognize and exert effector functions when challenged by pathogens that can enter the host. However, it was the description of pathogen recognition receptors (PRRs) and, particularly, of Toll-like receptors (TLRs) that led to understanding the prominent role that innate immunity plays in the recognition of microbial pathogens and in the regulation of the immune system1.
One of the most interesting aspects of innate immunity is the wide range of molecules recognized by PRRs. These receptors recognize diverse structural components or ligands (Table 1)2, essential for the survival of microorganisms, known as pathogen-associated molecular patterns (PAMPs)2. Recognition of PAMPs by PRRs stimulates an intracellular signaling cascade that involves the activation of diverse transcriptional factors involved in regulating expression of cytokines. Such cytokines play important roles in host protection, in the activation and migration of antigen presenting cells, and in the induction of the adaptive immune response.
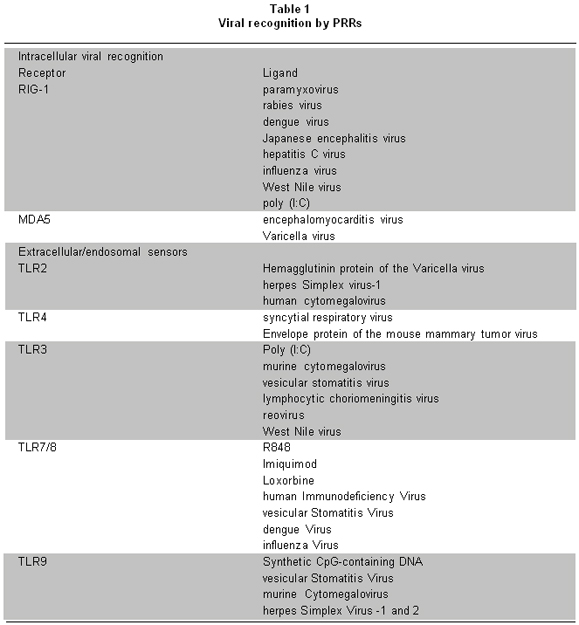
This review will focus on certain signaling PRRs that are potent triggers of inflammatory responses and play a very important role in viral infections. Among them there are the TLRs that can be either endosomal or extracellular3,4, and retinoic acid-inducible gene- (RIG-) I/MDA5 (melanoma differentiation-associated gene 5), known as RNA helicase-like receptors (RLRs). Furthermore, Double-stranded RNA-dependent protein kinase (PKR), 2',5'-oligoadenylate synthetase (2-5OAS), and adenosine deaminase acting on RNA (ADAR), known as effector proteins, complement the function of PRRs1. All these proteins are the main sensors of viral components and induce proinflammatory cytokine expression or interferon (IFN) response factors (Figure 1). This review discusses recent new knowledge on the molecular mechanism and role of PRRs in the recognition of viral PAMPs, the signaling pathways activated by such recognition, as well as the strategies used by viruses to evade such response.
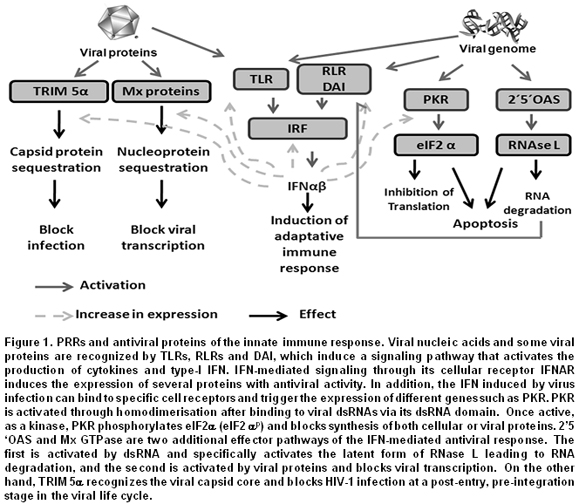
TLRs, adaptor molecules and their signaling pathways. TLRs are the best characterized members of the PRR family and can recognize both intracellular and extracellular pathogens4. Ten TLRs have been described in humans and all are type-1 membrane glycoproteins, from which six (TLR2, 3, 4 7/8, and 9) recognize viral components (Table 1). Some TLRs form hetero (TLR2/TLR1, TLR2/TLR6) or homodimers5. TLRs have an ectodomain or extracellular domain with leucine-rich repeats (LRRs), a transmembrane region and a cytoplasmic domain, homologous to the interleukin-1 (IL-1) receptor, known as Toll/IL-1R (TIR)3.
Briefly, the signaling pathway induced by TLRs is as follows: each TLR is stimulated by its specific ligand, which is recognized through the ectodomain. This leads to the recruitment, through the TIR, of adaptor proteins like the myeloid differentiation primary response protein 88 (MyD88), the Toll/IL-1 receptor domain containing adaptor protein (TIRAP), the Toll/IL-1 receptor domain containing adaptor inducing interferon-beta (TRIF), and the TRIF-related adaptor molecule (TRAM) (Figure 2). The IL-1R associated protein (IRAK) and tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) then also participate in the signaling pathway. TRAF6 acts as an ubiquitin ligase E and activates the transforming growth factor (TGF) b-activated kinase 1 (TAK1) complex and its subunits the TAK1-binding protein-(TAB) 1 and TAB2/3, responsible for the phosphorylation of NF-kappaB essential modulator (NEMO), which results in the activation of the kinase complexes constituted by inhibitor of kappaB (IkB) and IkappaB kinase (IkK) and in the degradation of phosphorylated IkB6. Finally, the nuclear factor kB (NF-kB), the activator protein 1 (AP-1), and the IFN regulatory factors (IRFs) are released and translocated to the nucleus where regulate the expression of molecules that participate in the inflammatory response and in the initiation of innate immunity4,7 (Figure 2).
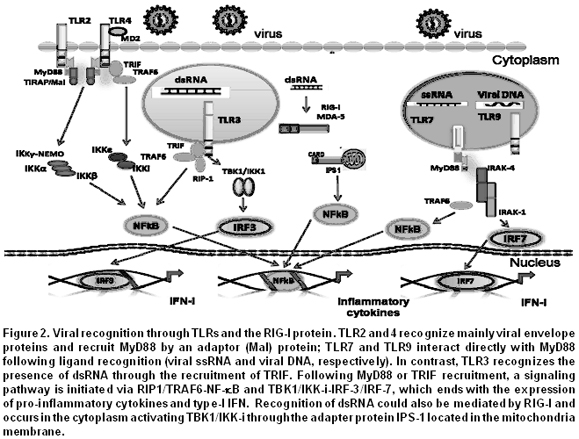
The stimulation of TLR7 and TLR9 with their respective ligand results in the formation of a complex constituted by MyD88, IRAK-4, TRAF6, TRAF3, IRAK-1, IKK-k, and IRF7 responsible for the activation of IRF7, which once phosphorylated is translocated to the nucleus and regulates the expression of type-I IFN. TLR3, through TRIF as adaptor protein, can induce the phosphorylation of IRF3 translocated to the nucleus and activates NF-kB and the expression of type-I IFN, as described above4,6,7.
In summary, the stimulation of TLRs through viral PAMPs or another origin allows the recruitment of adaptor proteins responsible for activating the induction of signaling pathways that result in the activation of transcriptional factors involved in the activation of the expression of genes whose products are involved in antiviral responses.
Recognition of viral components through TLRs. TLRs are components of a PAMP recognition system that act in concordance with other cellular factors to establish a bridge between the innate response and the adaptive immunity8. Their activation is initiated by a signaling pathway that leads to the activation of some mitogen-activated protein kinases (MAPKs) and of transcription factors like AP-1, IRF, and NF-kB. These transcription factors are responsible for the activation of genes that encode pro-inflammatory cytokines, such as the tumor necrosis factor a (TNF-a), interleukins (IL-1, 6, 8, and 18), IFN-a/b, chemokines, leukotrienes, class II major histocompatibility molecules and co-stimulatory molecules like CD40, CD80, and CD86, necessary for adequate antigenic presentation.
Initially, it was believed that the only TLRs involved in recognition of viral PAMPs were those recognizing nucleic acids like TLR3, TLR7/8, and TLR9, located in acid endosomes. However, certain authors and our unpublished results suggest that TLR2 and TLR4, located on the cell surface and known to recognize bacterial components, are also able to recognize viral PAMPs, mainly envelope glycoproteins9,10. A study carried out using TLR2 and MyD88-deficient mice infected with Lymphocytic choriomeningitis virus (LCMV) demonstrated that the TLR2-MyD88 interaction is essential for the activation of astrocytes and microglia and for the production of TNF-a and the monocyte chemo-attractant protein-1 (MCP-1)11. TLR2 also recognizes PAMPs from measles virus (MV), hepatitis C virus (HCV) and human herpes simplex virus type-1 and 2 (HSV1/2)12-14. In the case of HCV, it was demonstrated that an increase in the level of expression of TLR2 in individuals with chronic hepatitis is related to an increase in the level of circulating TNF-a and to the development of hepatic lesions13. A study performed in HEK293 cells stimulated with Epstein-Barr virus (EBV) showed that TLR2 is stimulated through NF-kB14,15. Acute infection by varicella zoster virus (VZV) is characterized by the development of an inflammatory response due to high cytokine production, specifically IL-6 and IL-8, which is associated with increased expression of TLR2 in human monocytes exposed to the virus16.
TLR4 is stimulated by bacterial lipopolysaccharides (LPSs) and endogenous ligands that are inflammation products, such as fibrinogen, fibronectin, b-defensins, and heat shock proteins7. Studies performed using TLR4-deficient mice infected with vesicular stomatitis virus (VSV) showed decreased expression of IL-12 and a higher viral load in comparison to mice expressing this receptor17. The role of TLRs in modulating the replication of certain viruses has also been examined. Using mice transgenic for Hepatitis B virus and intravenously injected with specific ligands for TLR2, 3, 4, 5, 7, and 9 showed that all receptors, except TLR2, can inhibit virus replication in IFN-dependent manner18. In summary, TLR4 is stimulated by PAMPs from the respiratory syncytial virus (RSV), coxsackie B virus, HSV, and MV19-21. However, additional studies must be pursued to determine the role of TLR2 and TLR4 in viral infections and in antiviral defense.
All endosomal TLRs are activated by nucleic acids. They also promote transcription of several cytokine and chemokine coding genes through the activation of NF-kB and MAPK, resulting in the expression of pro-inflammatory cytokines6. The main ligands of TLR3 are double-stranded RNA (dsRNA) genomes, and dsRNA replication intermediates of single-stranded RNA (ssRNA) viruses like RSV, dengue virus, and encephalomyocarditis virus22. TLR3 is also activated by contact with synthetic molecules or viral dsRNA analogs [poly (I:C)]. The final product of TLR3 activation is the induction of a strong inflammatory response characterized by the secretion of IL-12, TNF-a, IL-6, CXCL-10, IL-10, and IFN-a.
The role of TLR3 in antiviral immune response was demonstrated in TLR3-deficient mice, which are more susceptible to murine cytomegalovirus (MCMV), given lower expression of IFN23. TLR3-deficient mice infected with West Nile virus (WNV) develop higher viral loads24; however, the mice are less susceptible to developing meningitis, suggesting that the entry of the virus to the brain, and its pathogenesis, could be mediated by TLR3 activation and the immune response developed upon viral infection. TLR7 is also involved in antiviral responses. In humans, TLR7 is mainly expressed by plasmacytoid dendritic cells (pDCs), which after stimulation, induce the activation of NF-kb and MAPK and trigger the expression of cytokines such as IL-6, IL-12 and IFN-a, and the activation of co-stimulatory molecules through the TLR7 pathway15.
TLR7 and TLR8 are tightly related endosomal receptors and their ligands are ssRNAs. Both receptors are also activated by guanine analogs and uridine or guanosine-rich ssRNAs of either viral or cellular origin. Both receptors are crucial for the development of adaptive immune responses during viral infections, as is the case with influenza virus and dengue virus15,25.
Finally, the endosomal TLR9 is activated by at least three types of ligands with different biological outcomes26: i) viral DNA, ii) conventional CpG-containing DNA that activates B lymphocytes and induces the production of inflammatory cytokines by macrophages, and iii) D/A-type CpG containing-DNA that also stimulates the production of cytokines by macrophages and B lymphocytes, besides stimulating high production of type-I IFN by pDCs. It has been reported that this is the mechanism used by pDCs to respond through TLR9 to the presence of DNA viruses, such as adenoviruses, HSV1/2 and MCMV27-29. However, besides the recognition of DNA by TLR9, a different TLR9-independent mechanism has been proposed allowing the recognition of dsDNA in the cytoplasm of macrophages and in non-immune cells, but also inducing activation of IRF3, NF-kb, and IFN-a30.
Together, this shows that upon stimulation by nucleic acids or viral proteins, TLRs induce the expression of pro-inflammatory cytokines, whose function is to protect the host from viral infection. Thus, type-I IFN is one of the main components of innate immunity and is regulated by signals initiated at both intracellular and extracellular levels.
RLRs, adaptor molecules and their signaling pathways. The RIG-I and the MAD5 are two RLRs that recognize, respectively, short dsRNAs with 5 triphosphate ends and long dsRNAs from viral genomic RNAs. Both proteins are cytoplasmic and members of the DExD/Hbox RNA helicases that are stimulated by dsRNAs. RIG-I/MDA5 have two recruiting domains: an RNA helicase domain and a caspase recruitment domain (CARD; also known as caspase recruiting domains). The first domain is responsible for the recognition of dsRNAs and for the recruitment of various proteins that activate signaling pathways, and the CARD domain recruits proteins implicated in type-I IFN expression31. An adaptor protein has been described in this new signaling pathway. It is known as IFN-a promoter stimulator (IPS-1), [or anti-viral signaling protein (MAVS), virus-induced signaling adaptor (VISA) or CARD adaptor inducing IFN-a (Cardif)], and when it interacts with the CARD domain, allows the recruitment of dsRNA, which results in the expression of type-I IFN due to the activation and translocation of IRF3, IRF7 and NF-kB to the nucleus32,33. RIG-I is up or down regulated by the ubiquitin ligase TRIM25 and by RNF125. This means that RIG-I/MDA5 recognizes viral dsRNAs in the cytoplasm, while TLR3 does so in the endosomes.
Recognition of viral components by RIG-I/MAD5: RNA helicase-like receptors (RLRs). Certain viral dsRNAs synthesized in the cytoplasm as viral life cycle intermediaries, or as part of viral genomes are not only recognized by TLR3, but also by RLRs such as RIG-I and MAD5, which induce the expression of type-I IFN through a signaling pathway independent of the signaling activated by TLR3. Working with RIG-I- (RIG-I-/) and MAD5- (MAD5-/-) deficient mice, it was demonstrated that RIG-I is essential in recognizing RNAs from paramyxoviruses, influenza virus, and Japanese encephalitis virus; MAD5 is essential in recognizing picornavirus RNA and for the expression of IFN34-36. These results show that RLRs recognize different types of viral RNAs and activate a signaling pathway involved in the expression of IFN, different from the pathway used by TLRs. It was recently shown that cells over-expressing RIG-I and stimulated by Newcastle disease virus (NDV) or VSV present increased levels of expression of type-I IFN; in contrast, cells with low-level expression of RIG-I and stimulated by NDV, VSV, Sendai virus, HCV, or WNV show decreased levels of IFN37.
Because RIG-I/MAD5a induces antiviral responses, it has been suggested that the signaling pathways of both proteins have a common adaptor protein, but it is unclear whether they act in synergy when recognizing viruses. However, MDA5 not only participates in antiviral responses, it also inhibits the growth of tumor cells, through type-I IFN34,35.
Effector proteins involved in viral PAMP recognition. In the previous sections, the main PRRs involved in the induction of antiviral responses upon exposure to viral PAMPs were described. Nevertheless, it is also worth mentioning some proteins encoded by IFN-stimulated genes (ISGs). These proteins recognize virus structural components and through diverse mechanisms can activate antiviral responses. The first effector protein described with such characteristics was the PKR, which is an IFN-inducible serine-threonine kinase, but in contrast to PRRs, its activation does not regulate transcription, but blocks protein synthesis38. PKR possesses two dsRNA-binding motifs (dsRBMs) in its N-terminal region and a kinase motif in its C-terminal region. As a monomer, PKR is inactive, but its binding with dsRNA or highly structured RNAs induces its homodimerization and stimulates its autophosphorylation and auto-activation39. Once in the active state, PKR is dissociated from the dsRNA and phosphorylates the a-subunit of the eukaryotic translation initiation factor 2 (eIF2a), resulting in the inhibition of cellular and viral protein synthesis. PKR can also be stimulated by pro-inflammatory factors, such as growth factors, cytokines38, or the PKR-activating protein (PACT). In virus infected cells, most of the natural PKR activators are viral dsRNAs or viral life cycle replication intermediates. However, complex viruses with DNA genomes like Vaccinia virus (VV), Adenovirus, HSV-1, or HSV-2 have open reading frames (ORFs) in opposite orientation. This allows the production of overlapping mRNA transcripts that result in the production of dsRNAs that can in turn activate PKR. Interfering RNA (RNAi) used to silence PKR, or PKR knockout mice, has shown a reduction in their expression affects IFN production upon WNV infection, making the mice more susceptible40,41.
The presence of viral RNA also activates 2-5OAS, also known as 25 A synthetase. This enzyme is activated by IFN and was initially described as regulator of IFN synthesis, and, hence, as a key factor in antiviral responses40,42. 2´5´OAS promotes the degradation of viral RNA and catalyzes the synthesis of 25 oligadenylates that activate the latent cellular RNase (RNAse L)42.
ADAR1, 2, and 3 belong to another family of proteins known as adenosine deaminase acting on RNA (ADAR). ADAR1 is induced in response to IFN and has two different isoforms: ADAR1p150 and ADAR1p110. Recently, it was described that, in infected cells, dengue virus 2 (strain TSV01) activates the expression of genes regulated by IFN, such as ADAR, PKR, and 25OAS43. It has also been demonstrated that ADAR1 is responsible for editing the Hepatitis delta virus RNA, independently of IFN stimulation44. However, one of the most interesting aspects of ADAR is its participation in the biogenesis of microRNA, which are non coding RNAs implicated in the regulation of the expression of genes, whose products participate in various cellular processes. Additionally, it has been reported that ADAR1-induced modifications are necessary for the maturation of such RNAs45.
Recently, the stimulator of interferon gene (STING) protein, a new adaptor molecule that induces the expression of type-I IFN and plays a role in antiviral response was described. STING has 5 possible transmembrane domains; if STING is present in the endoplasmic reticulum, it can activate both NF-kb and IRF3 and induce the expression of type-I IFN46,47. Studies made with mouse embryonic fibroblasts that do not express STING show that such cells are susceptible to infection by viruses with negative polarity RNAs, including VSV48.
Another mechanism of host-virus recognition is through the Fv [or tripartite motif (TRIM)] protein. Many proteins with such motifs have been described. The different isoforms are produced by alternative mRNA splicing and each variant codes for a unique domain in its C-terminus. TRIM proteins, including TRIM5a that has in its C-terminal region the B30.2 or SPRY motif, can down regulate replication of retroviruses, such as HIV-1 and Murine leukemia virus49. On the other hand, some proteins members of the GTPase family, known as MX, are involved in recognition of viral proteins like the nucleocapsid, which after being recognized change their cellular location, affecting the production of new viral particles.
All these mechanisms demonstrate the existence of a wide variety of strategies for pathogen recognition and host protection upon viral infections. In summary, effector proteins play antiviral roles through 4 different mechanisms: i) arrest of protein synthesis, ii) degradation of viral RNA, iii) adenosine deaminase enzymes play an important role in converting adenosines to inosines (AàI), inducing errors in translation, and iv) recognition of viral proteins. However, some of these processes can drastically affect cell viability and cause the induction of apoptosis43. For this reason, the antiviral response induced by these types of proteins is immediate and observed only during the early stages of the infection, before the activation of the adaptive immune response.
Role of TLRs in viral pathogenesis. As described in this review, the aim of the recognition of viral PAMPs by PRRs is to control infection and/or contribute to the development of the adaptive immune response. However, prolonged activation of the innate immunity by TLRs can contribute to viral pathogenesis; for example, in WNV-infected mice in which the activation of TLR3 induces elevated production of TNF-a and IL-6 involved in the development of an inflammatory state, the permeabilization of the hematoencephalic barrier is induced, allowing viral entry and resulting in greater infection of the central nervous system24. Similarly, it has been described that RSV induces the over-expression of TLR3 and TLR4 that trigger an inflammatory state in the respiratory mucosa of the infected individual, who is rendered vulnerable to infections by other pathogens19.
In cells stimulated in vitro with specific TLR2, 3, 4, 5, 7, 8, and 9 ligands and infected with HIV-1, both naïve and memory T CD4+ and T CD8+ lymphocytes are activated. T CD8+ lymphocytes are activated and begin to express CD69, which promotes their own retention in lymphoid tissues, but T CD4+ cells lose the ability to express CD69. Despite these phenotypic changes, the cell population enters the cell cycle, but grows poorly, and can even undergo apoptosis. These results suggest that the systemic activation of TLRs through diverse ligands favors immune activation, effector cell sequestering, and T cell turnover. All these events can contribute to the immune dysfunction caused by HIV-1 and to the loss of T CD4+ lymphocytes in chronic HV-1 infections50.
Applications of TLRs in immunotherapies for viral disease control. Because TLRs are important for the induction of the innate immune response, understanding the molecular mechanisms involved in their activation could contribute to the development of immune therapies of vaccines, not only for viral diseases, but also for sepsis, allergies, autoimmune diseases, and cancer. TLR activation induces several different effects in immune cells, such as cytokine production, the positive regulation of co-stimulatory molecules like CD40, CD80, and CD86; for pDCs, which when stimulated through TLR9, induce IFN-a production. In this instance, besides the antiviral effect of IFN, it also influences the transport and clustering of pDCs51. This phenomenon is indispensable for the stimulation of the adaptive immune response in lymph nodes51.
Other studies also carried out with pDCs have demonstrated that early stimulation (4 h post-stimulation) with TLR7 agonists activates the antiviral cell machinery. Furthermore, stimulation of TLR7 and TLR8 with imiquimod and resiquimod, specific agonist, respectively, have therapeutic effects in basal cell carcinoma, genital lesions, and other epithelial lesions, generally associated with chronic human papillomavirus (HPV) infections52; these compounds induce apoptosis of HPV-infected cells and of other epithelial cells with dysplasic or neoplasic changes.
Evasion of innate immunity by viruses. As a consequence of the interaction between PRRs and PAMPs, the signaling pathways activated result in cytokine and IFN expression, or in the case of effector proteins, in inhibition of protein synthesis or degradation of viral dsRNAs. However, viruses has developed strategies to evade or take advantage of such immunological barriers for productive infections. Among the strategies already described is blockage of certain steps in the synthesis of IFN-a/b, inactivation of secreted IFN molecules, interference with signaling and/or blockage of the activity of effector antiviral proteins through their sequestration, production of viral homologous proteins, or competition. In the following paragraphs, we briefly describe some of the cell components manipulated by viruses to evade the innate immune response.
Expression of type-I IFN depends on the activation of IRF3 and IRF7 via IKK epsilon and TBK1. The genome of Rabies virus, Borna disease virus, and Ebola virus code for the P phosphoprotein and VP35 that can block the antiviral response induced by IFN53-55. In contrast, the human herpes simplex virus 8 encodes different analogs of IRF with negative dominant activity, allowing it to interfere with the activity of cellular IRFs56. The infected cell polypeptide 0 (ICP0) from Bovine herpes virus can interact with IRF3 and induce its proteasome-dependent degradation57. Similarly, the V protein of paramyxoviruses interacts with MD5-a and inhibits IFN-a expression58.
The genome of VV encodes two proteins (A46 and A52) that specifically inhibit the TLR-dependent signaling pathway. A46 interacts with the adaptor MyD88, TRIF and TRAM; while A52 interacts with TRAF and IRAK-2, inhibiting the formation of the complex implicated in the signaling pathway59. During chronic infections, the NS3/4A protease of HCV degrades the TRIF adaptor of TLR3, resulting in an alteration of the antiviral response induced by dsRNA. There is also evidence regarding inhibition of PKR by viruses via several pathways: VV E3L, Influenza virus NS1, and Reovirus d3 proteins can sequester dsRNAs and prevent PKR activation. Other viruses express dsRNA or highly structured RNAs that compete for binding to PKR. EBER-1 and EBER-2, expressed during the latent state of EBV and HCV IRES are some such examples40. Inhibition of PKR can also result from protein-protein interactions, as is the case of the HCV NS5 and VV E3L that interact with PKR and inhibit its function40,41,55.
The transcription factor NF-kb is key in the induction of the IFN expression and of pro-inflammatory cytokines involved in antiviral response. However, it can also be beneficial for viruses, given that the promoters of some viruses, including HIV-1, CMV, and EBV have binding sites for NF-kb, which facilitates transcription of the viral genome. Hence, some viruses can up- or down-regulate the NF-kkb expression; for example, some proteins of the African swine fever virus are able to regulate its expression. In the early stages of infection, the early viral protein A238L inhibits NF-kb expression, but once the infection progresses the late viral protein A224L stimulates its expression60. Furthermore, besides the direct strategies inhibiting IFN expression, VV expresses the A52R protein, which contains a TIR domain that interacts with the cytoplasmic proteins IRAK-2 and TRAF6 involved in TLR signaling59. Consequently, VV can affect the signaling induced by TLRs and additionally reduce the extent of the antiviral immune response.
CONCLUSIONS
The strategies used by vertebrates to recognize viruses are similar to those used to recognize other microorganisms, but for the former, the main PAMPs are usually nucleic acids or glycoproteins. As a result, screening of these molecules is performed in the same cell compartment as described for the PAMPs of other pathogenic agents. From this perspective, compartmentalization of the innate system has some similarity with the adaptive immune system because both systems use certain factors, depending on their cellular location. It is clear; however, as shown here, that upon infection, the immune response is initiated with the recognition of viral PAMPs by PRRs, resulting in the up-regulation of IFN-a/b. Since the discovery of IFN, 50 years ago, knowledge about the signaling pathways that regulate its production has greatly expanded. The discovery of different PRRs has led to better understanding the interactions between the innate and adaptive immune responses and has favored the development of new therapies owing to the discovery that the stimulation of TLRs plays an important role in the protection from, or in the development of diseases. This highlights the importance of studies aiming to better understand the regulatory mechanisms and the signaling pathways associated with the PRRs in response to PAMPs.
Other molecules that have also gained importance are the effector proteins, which inhibit protein synthesis, degrade RNA, or modify viral genomes and are also involved in launching the activation of the innate immune system and in developing the adaptive immune response.
ACKNOWLEDGEMENTS
This work was funded by Colciencias through grant 111540820517-2. Silvia Torres is a recipient of a Colciencias PhD scholarship.
Conflict of interest. None of the authors has conflicts of interest related to this study.
REFERENCES
1. Janeway CA, Jr., Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002; 20: 197-216. [ Links ]
2. Palm NW, Medzhitov R. Pattern recognition receptors and control of adaptive immunity. Immunol Rev. 2009; 227: 221-33. [ Links ]
3. Janeway C, Jr., Medzhitov R. Viral interference with IL-1 and toll signaling. Proc Natl Acad Sci USA. 2000; 97: 10682-3. [ Links ]
4. Netea MG, van der Graaf C, Van der Meer JW, Kullberg BJ. Toll-like receptors and the host defense against microbial pathogens: bringing specificity to the innate-immune system. J Leukoc Biol. 2004; 75: 749-55. [ Links ]
5. Kang JY, Nan X, Jin MS, Youn SJ, Ryu YH, Mah S, et al., Recognition of lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6 heterodimer. Immunity. 2009; 31: 873-84. [ Links ]
6. Zhang G, Ghosh S. Toll-like receptor-mediated NF-kappaB activation: a phylogenetically conserved paradigm in innate immunity. J Clin Invest. 2001; 107: 13-9. [ Links ]
7. Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol. 2004; 4: 499-511. [ Links ]
8. Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett. 2003; 85: 85-95. [ Links ]
9. Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, et al., Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol. 2003; 77: 4588-96. [ Links ]
10. Sato A, Linehan MM, Iwasaki A. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc Natl Acad Sci U S A. 2006; 103: 17343-8. [ Links ]
11. Zhou S, Kurt-Jones EA, Mandell L, Cerny A, Chan M, Golenbock DT, et al., MyD88 is critical for the development of innate and adaptive immunity during acute lymphocytic choriomeningitis virus infection. Eur J Immunol. 2005; 35: 822-30. [ Links ]
12. Bieback K, Lien E, Klagge IM, Avota E, Schneider-Schaulies J, Duprex WP, et al., Hemagglutinin protein of wild-type measles virus activates toll-like receptor 2 signaling. J Virol. 2002; 76: 8729-36. [ Links ]
13. Shehata MA, Abou El-Enein A, El-Sharnouby GA. Significance of toll-like receptors 2 and 4 mRNA expression in chronic hepatitis C virus infection. Egypt J Immunol. 2006; 13: 141-52. [ Links ]
14. Gaudreault E, Fiola S, Olivier M, Gosselin J. Epstein-Barr virus induces MCP-1 secretion by human monocytes via TLR2. J Virol. 2007; 81: 8016-24. [ Links ]
15. Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, et al., Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A. 2004; 101: 5598-603. [ Links ]
16. Wang JP, Kurt-Jones EA, Shin OS, Manchak MD, Levin MJ, Finberg RW. Varicella-zoster virus activates inflammatory cytokines in human monocytes and macrophages via Toll-like receptor 2. J Virol. 2005; 79: 12658-66. [ Links ]
17. Haynes LM, Moore DD, Kurt-Jones EA, Finberg RW, Anderson LJ, Tripp RA. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J Virol. 2001; 75: 10730-7. [ Links ]
18. Isogawa M, Robek MD, Furuichi Y, Chisari FV. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J Virol. 2005; 79: 7269-72. [ Links ]
19. Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, Tripp RA, et al., Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000; 1: 398-401. [ Links ]
20. Fairweather D, Yusung S, Frisancho S, Barrett M, Gatewood S, Steele R, et al., IL-12 receptor beta 1 and Toll-like receptor 4 increase IL-1 beta- and IL-18-associated myocarditis and coxsackievirus replication. J Immunol. 2003; 170: 4731-7. [ Links ]
21. Hahm B, Cho JH, Oldstone MB. Measles virus-dendritic cell interaction via SLAM inhibits innate immunity: selective signaling through TLR4 but not other TLRs mediates suppression of IL-12 synthesis. Virology. 2007; 358: 251-7. [ Links ]
22. Tsai YT, Chang SY, Lee CN, Kao CL. Human TLR3 recognizes dengue virus and modulates viral replication in vitro. Cell Microbiol. 2008; 11: 604-615. [ Links ]
23. Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, et al., Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci U S A. 2004; 101: 3516-21. [ Links ]
24. Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004; 10: 1366-73. [ Links ]
25. Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196-200. [ Links ]
25. Wang JP, Liu P, LAtz E, Golenbock DT, Finberg RT, Libraty DH. Flavivirus Activation of Plasmacytoid Dendritic Cells Delineates Key Elements of TLR7 Signaling beyond Endosomal Recognition. J Immunol. 2006; 177: 7114-7121 [ Links ]
26. Kumagai Y, Takeuchi, O., Akira, S. TLR9 as a key receptor for the recognition of DNA. Advanced drug delivery reviews. 2007; 60: 795-804. [ Links ]
27. Iacobelli-Martinez M, Nemerow GR. Preferential activation of Toll-like receptor nine by CD46-utilizing adenoviruses. J Virol. 2007; 81: 1305-12. [ Links ]
28. Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood. 2004; 103: 1433-7. [ Links ]
29. Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. 2003; 198: 513-20. [ Links ]
30. Ishii KJ, Akira S. Innate immune recognition of, and regulation by, DNA. Trends Immunol. 2006; 27: 525-32. [ Links ]
31. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al., Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006; 441: 101-5. [ Links ]
32. Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, et al., Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005; 23: 19-28. [ Links ]
33. Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, et al., Essential role of mda-5 in type-I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci USA. 2006; 103: 8459-64. [ Links ]
34. Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al., The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004; 5: 730-7. [ Links ]
35. Heim MH. RIG-I: an essential regulator of virus-induced interferon production. J Hepatol. 2005; 42: 431-3. [ Links ]
36. Hornung V, Ellegast, J., Kim, S., Brzozka, K., Jung, A., Kato, H. 5'-Triphosphate RNA is the ligand for RIG-I. Science. 2006 314: 994-7. [ Links ]
37. Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, et al., Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008; 82: 335-45. [ Links ]
38. Langland JO, Cameron, J.M., Heck, M.C., Jancovich, J.K., Jacobs, B.L. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006; 119: 100-10. [ Links ]
39. Williams BR. Signal integration via PKR. Sci STKE. 2001; 2001: RE2. [ Links ]
40. Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001; 14: 778-809, table of contents. [ Links ]
41. Langland JO, Cameron JM, Heck MC, Jancovich JK, Jacobs BL. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006; 119: 100-10. [ Links ]
42. Leroy MP, Baise EA, Pire GA, Desmecht DJ. Contribution of MX dynamin, oligoadenylate synthetase, and protein kinase R to anti-paramyxovirus activity of type 1 interferons in vitro. Am J Vet Res. 2007; 68: 988-94. [ Links ]
43. Umareddy I, Tang KF, Vasudevan SG, Devi S, Hibberd ML, Gu F. Dengue virus regulates type-I interferon signaling in a strain-dependent manner in human cell lines. J Gen Virol. 2008; 89: 3052-62. [ Links ]
44. Hartwig D, Schutte C, Warnecke J, Dorn I, Hennig H, Kirchner H, et al., The large form of ADAR 1 is responsible for enhanced hepatitis delta virus RNA editing in interferon-alpha-stimulated host cells. J Viral Hepat. 2006; 13: 150-7. [ Links ]
45. Faller M, Guo F. MicroRNA biogenesis: theres more than one way to skin a cat. Biochim Biophys Acta. 2008; 1779: 663-7. [ Links ]
46. Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al., The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008; 29: 538-50. [ Links ]
47. Nakhaei P, Hiscott J, Lin R. STINGing the antiviral pathway. J Mol Cell Biol. 2: 110-2. [ Links ]
48. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signaling. Nature. 2008; 455: 674-8. [ Links ]
49. Yap MW, Nisole S, Lynch C, Stoye JP. Trim5alpha protein restricts both HIV-1 and murine leukemia virus. Proc Natl Acad Sci USA. 2004; 101: 10786-91. [ Links ]
50. Funderburg N, Luciano AA, Jiang W, Rodriguez B, Sieg SF, Lederman MM. Toll-like receptor ligands induce human T cell activation and death, a model for HIV pathogenesis. PLoS One. 2008; 3: e1915. [ Links ]
51. Asselin-Paturel C, Trinchieri G. Production of type-I interferons: plasmacytoid dendritic cells and beyond. J Exp Med. 2005; 202: 461-5. [ Links ]
52. Miller RL, Meng TC, Tomai MA. The antiviral activity of Toll-like receptor 7 and 7/8 agonists. Drug News Perspect. 2008; 21: 69-87. [ Links ]
53. Brzozka K, Finke S, Conzelmann KK. Identification of the rabies virus alpha/beta interferon antagonist: phosphoprotein P interferes with phosphorylation of interferon regulatory factor 3. J Virol. 2005; 79: 7673-81. [ Links ]
54.Conzelmann KK. Transcriptional activation of alpha/beta interferon genes: interference by nonsegmented negative-strand RNA viruses. J Virol. 2005; 79: 5241-8. [ Links ]
55. Feng Z, Cerveny M, Yan Z, He B. The VP35 protein of Ebola virus inhibits the antiviral effect mediated by double-stranded RNA-dependent protein kinase PKR. J Virol. 2007; 81: 182-92. [ Links ]
56. Lin R, Genin P, Mamane Y, Sgarbanti M, Battistini A, Harrington WJ, Jr., et al., HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene. 2001; 20: 800-11. [ Links ]
57. Saira K, Zhou Y, Jones C. The infected cell protein 0 encoded by bovine herpesvirus 1 (bICP0) induces degradation of interferon response factor 3 and, consequently, inhibits beta interferon promoter activity. J Virol. 2007; 81: 3077-86. [ Links ]
58. Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, et al., The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A. 2004; 101: 17264-9. [ Links ]
59. Bowie A, Kiss-Toth E, Symons JA, Smith GL, Dower SK, ONeill LA. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc Natl Acad Sci U S A. 2000; 97: 10162-7. [ Links ]
60. Tait SW, Reid EB, Greaves DR, Wileman TE, Powell PP. Mechanism of inactivation of NF-kappa B by a viral homologue of I kappa b alpha. Signal-induced release of i kappa b alpha results in binding of the viral homologue to NF-kappa B. J Biol Chem. 2000; 275: 34656-64. [ Links ]

