










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombia Médica
On-line version ISSN 1657-9534
Colomb. Med. vol.42 no.1 Cali Jan./Mar. 2011
Genomic study of the critical region of chromosome 21 associated to Down syndrome
Julio César Montoya MSc1 , Juliana Soto, BSc.2, José María Satizábal, MD, MSc3, Adalberto Sánchez, PhD4, Felipe García, PhD5
1. Associate Professor, Department of Physiological Sciences, School of Basic Sciences, Faculty of Health, Universidad del Valle. Professor, Faculty of Basic Sciences, Universidad Autónoma de Occidente, Cali, Colombia. e-mail: jcmontoya@uao.edu.co
2. COLCIENCIAS Young Researcher, Laboratory of Molecular Biology and Pathogenesis, Department of Physiological Sciences, School of Basic Sciences, Faculty of Health, Universidad del Valle, Cali. e-mail: dendroapsis@gmail.com
3. Full Professor, Department of Physiological Sciences, School of Basic Sciences, Faculty of Health, Universidad del Valle, Cali, Colombia. e-mail: josemariasatizabal@yahoo.es
4. Associate Professor, Department of Physiological Sciences, School of Basic Sciences, Faculty of Health, Universidad del Valle, Cali, Colombia. e-mail: asanchez6911@yahoo.com
5. Full Professor and Scientific Director of the Laboratory of Molecular Biology and Pathogenesis, Department of Physiological Sciences, School of Basic Sciences, Faculty of Health, Universidad del Valle, Cali, Colombia. e-mail: labiomol@gmail.com
Received for publication February 16, 2010 Accepted for publication June 30, 2010
SUMMARY
Introduction: Previous reports have identified a region of chromosome 21 known as Down ayndrome critical region (DSCR) in which the expression of some genes would modulate the main clinical characteristics of this pathology. In this sense, there is currently limited information on the architecture of the DSCR associated.
Objective: To obtain in silico a detailed vision of the chromatin structure associated with the evaluation of genomic covariables contained in public data bases.
Methods: Taking as reference the information consigned in the National Center for Biotechnology Information, the Genome Browser from the University of California at Santa Cruz and from the HapMap project, a chromosome walk along 21 Mb of the distal portion of chromosome 21q arm was performed. In this distal portion, the number of single nucleotide polymorphisms (SNP), number of CpG islands, repetitive elements, recombination frequencies, and topographical state of that chromatin were recorded.
Results: The frequency of CpG islands and Ref genes increased in the more distal 1.2 Mb DSCR that contrast with those localized near to the centromere. The highest level of recombination calculated for women was registered in the 21q22.12 to 22.3 bands. DSCR 6 and 9 genes showed a high percentage of methylation in CpG islands in DNA from normal and trisomic fibroblasts. The DSCR2 gene exhibited high levels of open chromatin and also methylation in some lysine residues of the histone H3 as relevant characteristics.
Conclusion: The existence of a genomic environment characterized by high values of recombination frequencies and CpG methylation in DSCR 6 and 9 and also DSCR2 genes led us to postulate that in non-disjunction detected in Down syndrome, complex genomic, epigenetic and environmental relationships regulate some processes of meiosis.
Keywords: Down syndrome; Down syndrome critical region; Genomics; Chromatin architecture; Epigenetics; Non-disjunction.
Colomb Med. 2011; 42: 26-38
Estudio genómico de la región crítica del cromosoma 21 asociada con el síndrome de Down
RESUMEN
Introducción: Análisis previos han identificado una región del cromosoma 21, conocida como región crítica del síndrome de Down (DSCR) en donde se localizan algunos genes cuya expresión modularía las principales características clínicas de este síndrome. En este sentido, existe poca información detallada sobre la arquitectura de la cromatina asociada con la DSCR.
Objetivo: Obtener in silico, a partir de la evaluación de covariables genómicas contenidas en bases de datos públicas, una visión detallada de la estructura cromatina asociada con la DSCR.
Métodos: Tomando como referencia la información consignada en el National Center for Biotechnology Information, el Genome Browser de la Universidad de California en Santa Clara y el proyecto internacional HapMap, se efectuó un paseo cromosómico a lo largo de 21Mb de la porción distal del brazo q del cromosoma 21, para registrar el número de polimorfismos de nucleótido único, el de islas CpG, de secuencias repetidas, las tasas de recombinación y el estado topológico de la cromatina asociada.
Resultados: La frecuencia de islas CpG y de genes referenciados se incrementó en los últimos 1,2 Mb de la región distal en contraste con su distribución pericentromérica. La mayor tasa de recombinación calculada en este estudio para mujeres se registró en las bandas 21q22.13 y 21q22.3. Los genes DSCR 6 y 9 presentaron un elevado grado de metilación en islas CpG tanto en fibroblastos normales como en trisómicos. En el gen DSCR2 se observó un alto grado de descondensación cromatínica, además de metilación de diferentes residuos de lisina de la histona H3.
Conclusiones: La existencia de un ambiente genómico caracterizado por tener elevadas tasas de recombinación y de metilación de genes DSCR 6 y 9, permite postular que en la no disyunción asociada con el SD, operarían complejas interacciones genómicas, epigenéticas y ambientales que actuarían en algunos procesos meióticos.
Palabras clave: Síndrome de Down; Región crítica del síndrome de Down; Genómica; Arquitectura cromatínica; Epigenética; No disyunción.
Colomb Med. 2011; 42: 26-38
Down syndrome (DS) is a numerical chromosomal abnormality, consisting of an extra chromosome 21 and it is the most common genetic cause of mental retardation in humans; the incidence of DS is of 1/600 to 1/1000 live births1. In the city of Cali, a descriptive study carried out between 1991 and 1995 determined an incidence of 1.5 per 1000 live births with Down (1/650)2, a value similar to that registered in other studies1,3.
The embryological etiology of DS is mainly correlated with the non-disjunction of a chromosome 21 during la maternal meiosis4; this consists of errors occurring during the separation of homologous chromatid pairs. The other cases are associated to translocations (3%) or mosaicism (2%) generated by inherited or direct structural alterations of the cygote5. In spite of the numerous clinical and cytogenetic studies on DS, little is still known about its molecular origin; however, previous studies have shown that the phenomenon of the non-disjunction is associated to the alteration of the recombination mechanisms and to epigenetic processes of chromatin remodeling like methylation of the CpG islands of chromosome 216-8.
Comparisons of the triplicate regions of chromosome 21 among individuals with DS phenotype have permitted identifying a common overlap region that contains some of the candidate genes that could explain the pathologies associated to DS. This region is known as Down Syndrome Critical Region (DSCR) and it is localized in the distal terminal of the long arm of chromosome 21 (21q22)9. The size of this region, until 2000, was considered to be from 1.6 to 2.5 Mb9,10; however, recent studies have detected a duplication from 4.3 to 5Mb between bands 21q22.13-q22.2 of chromosome 21, which includes a new candidate gene for DS (DSCR1/RCAN1) localized in the distal region of the DSCR, associated with cardiac abnormalities11,12.
Results obtained from the human genome project and from other genomes have broadened the frontiers of knowledge on their structure and function while offering answers to many lingering questions in biology and biomedical sciences. Within this scenario, the use of information technology in biomedical research has increased significantly in the last three decades. Within this current of knowledge, medicine has been influenced by the amount of genomic and post-genomic data, which have permitted conducting many biomedical studies and therapies that could not be performed until the beginning of this century. Currently, there is a vast amount of genomic data, many of which are available on-line, which have enhanced progress of in silico biology; this trans-discipline permits simulating biological processes by using software and algorithms, complementing that obtained experimentally and increasing understanding of many processes operating in biological systems13.
According to the aforementioned, and starting from the results obtained from the human genome project, it has been possible to study the structural and functional characteristics of vast chromosomal regions through quantitative and qualitative analysis of genomic variables associated to genetic and acquired disease. Some of these variables are the Alu elements, which belong to the SINE family (Short Interspersed Nuclear Elements) and are sequences of 250-280 nucleotides present in 1,500,0009 copies dispersed throughout the genome; CpG islands that are chromosomal regions enriched by C-G dinucleotides associated to genome regulating zones containing bonding sites to transcription factors; the SNPs (Single Nucleotide Polymorphisms), which are variations in the DNA sequence affecting a sole nucleotide (adenine, thymine, cytosine, or guanine) of the genome13,14.
DNA packaging in nucleosomes and its arrangement on the chromatic strand, generates a series of restrictions in processes like replication, DNA injury repair, genetic recombination, and transcription regulation; thus, in the interphase nucleus, where these processes take place, chromatin undergoes transient changes that permit solving these impediments via very varied mechanisms, most of which are epigenetic14. The mechanisms involved in chromatin dynamics are complex and include multi-protein assemblies with acetyltransferase, deacetylase, methyltransferase, or kinase activities of some histones, which are controlled by regulation factors in precise regions of the genome15. Thus, the nuclear chromatin is remodeled to permit or hinder regulated gene expression. The genome project has also detected a large amount of functional modifications related to the chromatin structure, and this has permitted defining two types of regions in the genome: a) «active» domains, which would be the regions defined by high transcription levels, early replication, histone H3 acetylation, and demethylation of lysine 27 of histone H3 (H3K27)16; b) «inactive» domains, corresponding to regions of latent replication with scant transcriptional activity, low H3 acetylation and methylation of H3K27. The active domains are rich in transcription starter sites, CpG islands, and Alu repetitions, while the LINE 1 and LTR repetitions are over-represented in the inactive domains16. In this context, a genomic environment is defined as a chromatin zone with differential characteristics that includes repetitive sequences, CpG islands, Guanine-Cytosine content, gene density, among others used to study at the genome level epigenetic biological processes occurring as a result of an alteration of cellular homeostasis. The systematization of the information obtained from all these genomic variables along the chromatin segments is what is known as the «chromosome walk».
Given that there is little detailed information on the architecture of chromatin associated to DSCR, in this study, through an extensive biotechnology analysis, a series of co-genomic variables were evaluated, permitting the characterization of the topological state of the critical regions associated to DS that would be implicated in processes of non-disjunction and in epigenetic events. The results obtained contributed strong evidence that the non-disjunction would compromise extensive regions of the telomeric portion of the q arm of chromosome 21; these were characterized for having high-density zones of CpG islands, portions of chromatin sensitive to treatment with DNAase I, with a variable degree of methylation of some residues of lysines from the histone H3; besides having a high intensity of recombination in regions coinciding with a high degree of polymorphisms by deletions and insertions.
METHODS
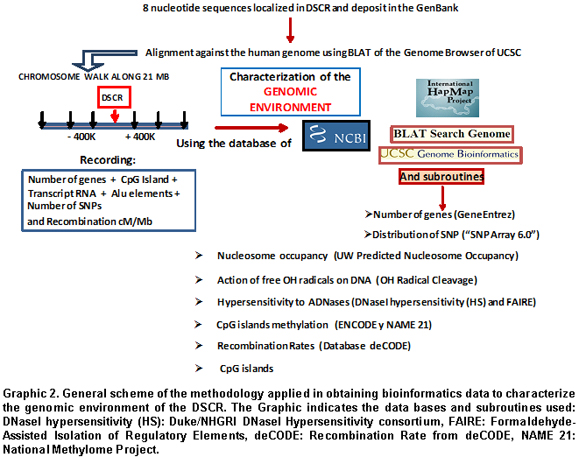
A descriptive bioinformatics study was conducted in which eight gene sequences were selected localized in the DSCR of chromosome 21 between bands 21q22.12 to 21q22.2. The sequences used for the study had been previously deposited in the GenBank (http://www.ncbi.nlm.nih.gov), with access codes: DSCR2: NM_605296, DSCR3: NM_006052, DSCR4. NM_005867, DSCR5: NR_028352, DSCR6: NM_018962.2, DSCR8: BC070229, DSCR9: NR_026719, DSCR10: NC_000 021. The data obtained from using the distinct bioinformatic platforms was gathered between August and December of 2009. For results where we used the genome Browser from the University of California at Santa Cruz, we applied the 2006 version.
Chromosome walk in the DSCR. Using bioinformatic tools available in the web pages for the United States National Center for Biotechnology Information (NCBI), the HapMap (www.hapmap.org), and the Genome Browser from the University of California at Santa Cruz (http://genome.ucsc.edu/), we conducted a chromosome walk in a 21-Mb extension (Graphic 1); this included ±8Mb adjacent to each of the DSCR genes; every 400Kb, we recorded the number of genes, CpG islands, transcribed RNA, Alu elements, the number of SNPs, and the recombination rates in cM/Mb. Additionally, we explored other characteristics like methylation of CpG islands, identification of methylation and acetylation sites, and sites hypersensitive to DNAses that would yield the base to define a genomic environment associated to the DSCR (Graphic 2).
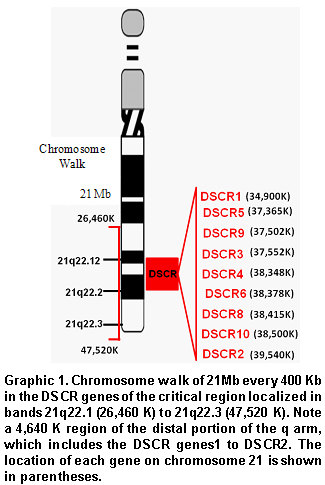
Values for the recombination rates for the DSCR, the pericentromeric and telomeric chromatin were obtained from the deCODE data base (recombination rate from deCODE, Marshfield, or Genethon Maps) (deCODE default) a subroutine of the genome Browser. Values obtained correspond to an analysis of 146 families where 1257 meiotic events were registered and discriminated by sex; for this work we only used those values registered for women.
G:C Content and distribution of CpG islands. To analyze data regarding the fine structure of the 21q22.12 to 21q22.2 region containing the DSCR (21q:34.909. 450-41.450.000), we used several subroutines of the Genome Browser. Data was obtained from corresponding bases; this was conducted by following the recommendations consigned for each of them. The G:C content was obtained by using the subroutine that calculates the G:C percentage in 5-pb windows along the analyzed region. The graphic representation of this program is a continuum of peaks and valleys, which permits -in any zone of the sequence- calculating the percentage in this window. To define and discriminate the distribution of free G:C from that included in zones of CpG islands, we used the -CpG Islands- subroutine («Islands <300 Bases are Light Green»); this predicts the existence of CpG islands from the search for dinucleotide G-C sequences, employing a score of +17 in a region of 200 pb. Scores over 0.6 indicate the existence of CpG islands. In addition, we explored the degree of methylation of these islands, using the data base of the ENCODE subroutine «HudsonAlpha Methyl-seq»; the program shows the average value of the state of methylation in CpG islands. The graphic visor identifies in orange those Methylated CpG islands (score of 1000), while blue is for non-Methylated CpG islands (score of 0).
Identification of structural alterations in DSCR. To identify SNP distribution, data consigned in the base collectively denominated simple nucleotide polymorphisms was obtained from the NCBI (http://www.ncbi.nlm.nih.gov/SNP) and complemented with those values from the 130- SNP 130- construct from the genome Browser. The graphic visor for this program discriminates the type of SNP by synonymous codon (green), non-synonymous codon (red), SNP non-translated (blue), and SNP in intron (black). Additionally, the SNP Array 6.0 was used, which includes more than 906,000 SNPs and 946,000 probes to detect the variation of the number of copies. To complete the information on the insertion and deletion polymorphisms in the DSCR, we obtained the values consigned in the data base from the National Human Genome Research Institute (NHGRI) (deletion/insertion polymorphisms in ENCODE regions) of the ENCODE project. Use of all the data was adjusted to the guidelines stipulated by the NHGRI Consortium (http://www.genome.gov/).
Structure of the chromatin associated with DSCR. Data on the structural and functional state of the chromatin associated with DSCR was obtained from the DNaseI hypersensitivity (HS) data bases from the Duke/NHGRI DNaseI Hypersensitivity consortium, and formaldehyde-assisted isolation of regulatory elements (FAIRE) from the University of North Carolina (UNC) included in the Genome Browser; these data were complemented with the values from the ChIP/Chromatin Immunoprecipitation to select regulating factors. For the analyses included in this work, we used the data from the high-resolution platforms 1% ENCODE tiled microarrays from NimbleGen by Roche (http://www.nimblegen.com/). Additionally, we used the Boston University - ORChiD (OH Radical Cleavage Intensity Database) subroutine from GBUCSC. This base provides prediction of cleavage intensity per nucleotide produced by the action of free radicals on DNA devoid of proteins in ENCODE regions. The graphic visor shows a structural profile from the DSCR DNA.
Additionally, we explored nucleosome occupancy by taking data from the uw predicted nucleosome occupancy subroutine contained in the Genome Browser. It contains the predictions of the other scores of nucleosome occupancy using experimental data from the A375 cellular line developed by Ozsolak et al.17 In the browser, the score corresponding to a 50-nucleotide oligo is assigned to the 26th base. A score of 0 indicates uncertainty in assigning the data, a score of 1.0 corresponds to a statistically reliable prediction, indicating frequent positions of nucleosome occupancy, and a score of -1.0 corresponds to a reliable prediction of low-density nucleosome areas.
Prediction of the degree of methylation in DSCR. Cytokine methylation, mainly in the CpG islands, is an indicator of transcriptional activity given that these elements are associated to active promoters. To determine the degrees of methylation of the DSCR genes, we employed the data obtained via the NAME 21 project18. The methylation patterns of chromosome 21 regions were analyzed for 190 gene promoters localized along this chromosome; using for this purpose, the bisulfite conversion and subsequent sequencing of subclones obtained from five different types of cells. Through this methodology, the methylation state from 580,427 CpG sites15 has been measured. For this study, we selected the data from normal fibroblasts and trisomic fibroblasts available for some of the DSCR genes.
Statistical analyses. All the statistical analyses were performed by using the STATISTICA 7 program (StataSoft Inc. NJ) and the Statgraphics program for Windows. We conducted a Kruskal-Wallis test to determine significant differences of the analyzed variables along the DSCR; a value of p<0.05 was considered significant. Through a multiple regression model, we assessed the correspondence between the recombination rates, number of SNPs, Alu elements, and genes in the eight DSCR genes analyzed.
RESULTS
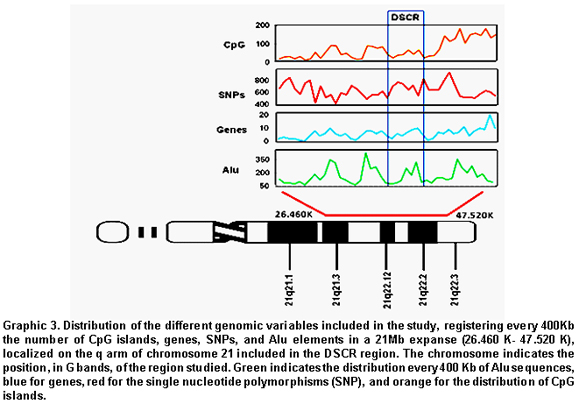
Distribution of different cogenomic variables in DSCR. The analysis conducted in a 21-Mb extension, which included all the critical regions reported, from the most centromeric (DCR1) to the most distal localized in a peritelomeric region (DCR2), demonstrated that the frequency of CpG islands increased in the last 1.2 Mb with a total of 468/3089 (15.2%); in contrast with the 2.7% (83/3089) of the total localized in the proximal region near the centromere. The gene distribution was similar to that registered for the CpG islands, where it was determined that 15.5% (39/252) was included in the terminal portion, while 2.8% (7/252) was localized in the proximal portion. Upon analyzing the frequencies of SNPs and Alu elements in the proximal and distal regions, statistically significant differences were not observed (p>0.05) (Graphic 3).
For the purpose of discriminating if the distribution of CpG islands, genes, and Alu elements was homogenous for each of the eight DSCR regions, we conducted a multiple regression analysis. This showed statistically significant differences for each of the DSCR genes (Graphic 4).
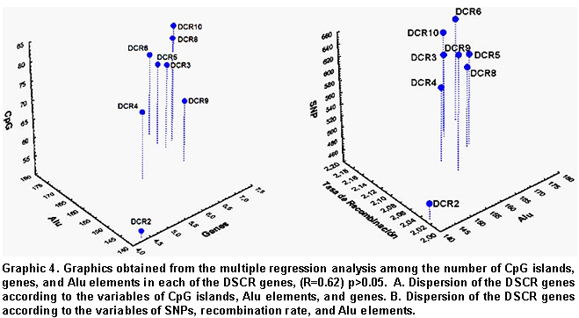
DSCR gene dispersion according to variables like CpG islands, Alu elements, and genes revealed greater correlation in DSCR10 and 8 genes and lower for DSCR2 (Graphic 4 A). Additionally, for variables like SNPs, rate of recombination, and Alu elements, it was determined that the greater correlation was observed in the DSCR10 and 6 genes, with the lower correlation coinciding for the DSCR2 gene (Graphic 4 B).
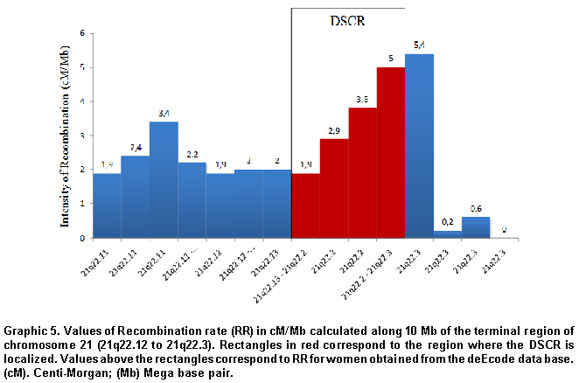
Recombination in DSCR. Taking as a base the recombination rate (RT) values consigned in the deCODE data base, an increase was noted in the recombination rate values within the loci 21q22.12 and 21q22.2, where part of the DSCR is localized (1.9 to 5 cM/Mb), in comparison with those proximal near to the centromere (1.9 to 2.0 cM/Mb) and in the telomeric (5.4 to 0 cM/Mb) (Graphic 5).
Characteristics of Chromatin Associated to DSCR. For each of the DSCR genes an analysis was performed including the most important characteristics of the structure of the portions of cellular chromatin they occupy (Graphic 6 A and 6 B).
The DSCR1 that extends between 34,909,450 to 34,909,749 localized in band 21q22.12 revealed that the G:C percentage is variable with an average of 47.5%; an extensive degree of methylation or acetylation was not determined on Lysines (K) of the Histone H3 in the cellular lines from which information was obtained (GM125 and K562). Toward the central region, occupancy was observed with a score of 0.75 and very low and discreet sensitivity to treatment with DNAaseI, Formaldehyde, and OH radicals.
The DSCR5 extending from 37,365,228 to 37,367,327 localized in the locus 21q22.13, presented an average C:G percentage of 56%; in this region a relative methylation was determined of the 3H K4me1 and me3 histone in GM125 cells, as well as acetylation in the same histone of the K27 residue moderate in the K562-cell chromatin. Toward the terminal portion of this gene, nucleosome occupancy was observed with an average score of 0.56. Extensive zones were not determined for sensitivity to DNAaseI, Formaldehyde, and treatment with OH radicals.
The DSCR9 extending from 37,502,267 to 37,514,691 and localized in band 21q22.13 was characterized because its average content of C:G was of 52%, with zones with poor methylation of the Histone H3 in GM125 cells besides acetylation of 3HK27ac in the first 700 pb of this gene; the region did not reveal sensitivity to treatments with DNAase I, Formaldehyde, and treatment with free radicals.
The DSCR3 was localized between 37,552,524 and 37,562,464 within the locus 21q22.13 where the average C:G percentage varied greatly reaching 62% toward the terminal region. Unlike the other genes, in this one a high degree of methylation was determined in 3H4K me1 and me3 in GM125 cells, besides acetylation of 3HK27ac in K562 cells. The chromatin associated to this gene did not present sites sensitive to standard treatments to determine fragility of the associated ADN; additionally, the nucleosome occupancy had an average score of 0.45 toward the terminal region.
The DSCR4 whose localization within the locus 21q22.13 extends from 38,348,250 to 38,415,286 showed an average C:G percentage of 48%; it is an H3 methylation zone toward the central and terminal portion, but which revealed a nucleosome occupancy score of 0.02, hypersensitive areas to treatments with DNAase I, formaldehyde and cleavage intensity by OH radicals.
The DSCR6 extending within the locus 21q22.13 (38.378.862 to 38.391.958) presented the highest average C:G percentage at 66%; with zones of extensive H3 methylation in the K27meG and K06meG positions; the nucleosome occupancy score was low with an average value of 0.2 besides the lack of areas hypersensitive to standard treatments used in this work.
The DSCR8 localized in band 21q22.13 (38.415.449 to 38.450.481) presented a very variable degree of C:G percentage; H3 K4me1,2,3 methylation in GM125 cells and modifications in 3HK27ac and 27me. It did not present a high score of nucleosome occupancy that was complemented with the lack of regions hypersensitive to treatment with DNAaseI, formaldehyde, and to the resistance to treatment with OH radicals.
The DSCR10 extending from 38.500.020 to 38.502.708 of the band 21q22.13, registered a mean C:G percentage of 58% with extensive H3 methylation and acetylation associated with a low score of nucleosome occupancy and some short zones with hypersensitivity to DNAaseI, formaldehyde, and resistance to treatment with OH radicals.
The DSCR2 is the closest to the telomere, extending within locus 21q22.2 of 39.477.821 to 39.478.113; it revealed an extensive segment of methylation of CpG islands not found in other genes. A high degree of H3 epigenetic modifications was determined with a nucleosome occupancy score of 0.2. It should be stressed that it is also a zone of high hypersensitivity to treatment with DNAaseI, formaldehyde and to treatment with OH radicals.
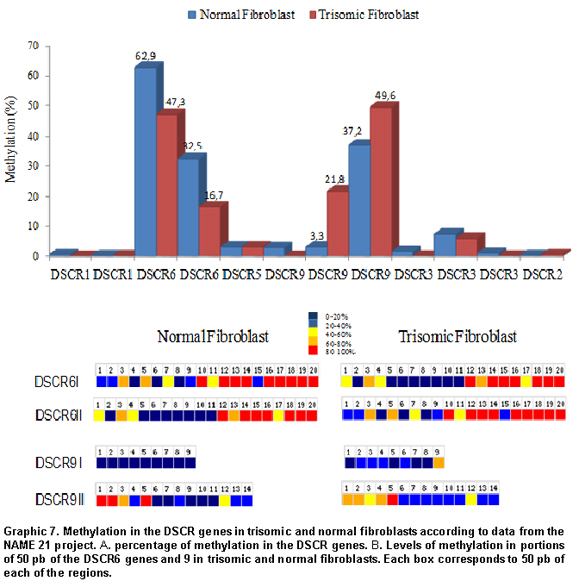
Methylation in the DSCR. Taking the data obtained from the chromosome 21NAME project as a point of departure, it was possible to explore the percentage of methylation of CpG islands in normal fibroblasts and compare it to that of trisomic fibroblasts. According to what is shown in Graphic 7A, the DSCR 6 and 9 genes had a high degree of CpG methylation in both types of cells. However, for the DSCR6, 62.9% methylation was registered in normal fibroblasts compared to 47.3% in trisomic fibroblasts. In the DSCR9, the trisomic fibroblasts had 49.6% methylation against 37.2% methylation in normal fibroblasts. A more detailed analysis of 50-bp portions of these regions, revealed statistically significant differences in the methylation styles for both loci in the two types of fibroblasts (Graphic 7B). The DSCR6 showed more methylation in 50-pb CpG portions in both types of cells in contrast with the same CpG distribution in the DSCR9.
DISCUSSION
One of the effects of the human genome project was the great volume of data consigned in different data bases that permitted their bioinformatics management. Results from these analyses have promoted a new way of understanding in silico biological phenomena, using different genomic variables consigned in on-line data bases. From this approach, 21 Mpb were explored from the terminal region of chromosome 21 including DSCR. In this regard, application of bioinformatic tools permitted analyzing the non-disjunction associated to recombination mechanisms and to epigenetic processes in which chromatin undergoes remodeling.
The results obtained revealed the existence of a heterogeneous structure of codifying sequences and non-codifying elements, which was asymmetric, increasing the number of CpG islands and genes toward the 21q22.12 to 21q22.2 region of the distal portion of the chromosome. Data obtained contributed to the knowledge of a differential genomic environment for the DSCR critical region.
Our data also showed that the distribution of the genomic variables included in this study (genes, CpG islands, SNPs, and Alu sequences), was different for each of the DSCR genes; with the DSCR2 gene showing greater variation. In this sense, it has been previously described that the DSCR2 is an important factor associated to DS, which is expressed in different tissues and fulfills key functions in regulating the function of chromosome 2119. The DSCR2 gene codifies a leucine-rich protein of approximately 288 amino acids and interacts with a nuclear receptor (PPARb)19.
A series of prior reports show that DS caused by meiotic non-disjunction of chromosome 21 is associated to advanced maternal age; however, even though this hypothesis is accepted in general terms, unanswered questions prevail regarding the role played by other associated risk factors5,12.
Prior studies suggest the existence of complex interactions between the effect of maternal age and recombination patterns altered during meiosis5,17. In this study, we noted that the recombination rate in the terminal region of the long arm of chromosome 21 (21q22.12 to 21q22.3) presented, for women, high recombination values, compared to those registered for men (decode data base). Furthermore, the recombination rate is lower in the 2Mb of the pericentromeric quarter and the most distal toward the telomere. Integrating these results with those obtained previously, it is possible to postulate that the region studied, would be a hot zone for recombination of portions of the chromatin associated to the DSCR.
Results obtained revealed that the terminal region of chromosome 21 (21q22.13) is relatively insensitive to treatment with DNAase I as with other agents, which permits determining that there are no extensive DNA unprotected zones in the DSCR. This fact is associated to the existence of a high degree of methylation and acetylation of several Lysines of the Histone H3, configuring a genomic environment that, via internal or external effectors, could alter the local recombination during meiosis. It has been identified that recombination alteration is correlated to non-disjunction both in human and animal models. Pericentromeric and telomeric exchanges are responsible for 34% of all the exchanges occurring in very young women compared to only 10% in adult older women7. In addition, it has been determined that chromatid exchange patterns in older women, are similar to those observed when non-disjunction of chromosomes 21 occurrs7.
In this study, another very relevant genomic factor was the degree of differential methylation observed between DSCR genes. The genes with higher degree of CpG methylation were DSCR 6 and 9 (Graphic 7). Prior studies have shown that CpG islands exert epigenetic control during embryonic development and are normally found in non-methylated state; however under some pathological situations, hypermethylation of a great portion of these islands is observed. Additionally, the methylation pattern would be correlated with the local structure of chromatin and its degree of repetition18,20.
A previous work revealed that the existence of susceptibilities in the exchange patterns is associated with the origin of the meiotic error and with age; the highest failures in meiosis I (MI) have been reported in younger mothers, progressively decreasing in the group of older mothers, which mimics the pattern observed in the disjunction of chromosome 2121. On the other hand, the association between the non-disjunction and recombination has been analyzed in a 5.1-Mpb telomeric region of chromosome 21. It was determined that the non-disjunction in telomeric regions occurred more frequently in the MI of oocytes of young mothers contrary to an increase of recombination errors in MII in a 5.7 Mpb pericentromeric portion, which were more frequent in the groups of older mothers22. It was previously determined that with increased age the loss of telomeric portions is more intense, which has permitted postulating the association between age and the terminal instability of the long arm of chromosome 21. Additionally, it has been proposed that not only the chronological age but the progressive loss of telomeric zones, becomes a differentiating factor among women young in age but old in telomeric terms. Thus, the telomeric wear of chromosome 21 is somehow associated to the non-disjunction in MI and MII and the subsequent risk of increased trisomic births in adult mothers23,24.
In conjunction with the results obtained by other authors, the greatest recombination rate determined in our study coincided with regions 21q22.13 to 22.2 of the distal region of chromosome 21 (Graphic 5), in chromatin areas associated to DSCR2 where we observed a high degree of chromatin decondensation and methylation of different Lysine residues of H3 in a way that the structure of the meiotic functional apparatus would correlate with the topologic and epigenetic state of the chromatin associated to DSCR where, as we have shown, chromatin undergoes structural remodeling, very possibly induced by fine epigenetic control. The failure in this chromatin remodeling, could be associated to previously reported patterns of non-disjunction in MI, which are more frequent in the same oocyte phase of young mothers.
At the functional level, it has been found that the expression levels of some of the genes localized in the critical region (DSCR4, DSCR6; KIR4.2, GIRK2, KCNE1 and KCNE2) are not over-expressed in the trisomic fetal brain, indicating that the DS phenotype cannot be simply explained by the effect of gene dosage imbalance. This is to other genetic, epigenetic factors also exist, along with environmental and stochastic events that destabilize the trisomic transcriptome, generating a series of metabolic alterations25. In summary, this work permitted confirmig and broadening the genomic and molecular knowledge of non-disjunction in DS via open-access bioinformatic tools. These results expand the frontiers of knowledge by introducing the in silico approach as a complement of clinical and experimental studies in this syndrome and in other pathologies.
ACKNOWLEDGMENTS
We thank Universidad del Valle and Universidad Autónoma for their financial support and for the infrastructure to conduct this research.
Conflict of interest. None of the authors has conflicts of interest related to this study.
REFERENCES
1. CanWeld MA, Honein MA, Yuskiv N, Xing J, Mai CT, Collins JS, et al. National estimates and race/ethnic-specific variation of selected birth defects in the United States, 1999-2001. Birth Defects Res A Clin Mol Teratol. 2006; 76: 747-56. [ Links ]
2. Ramírez RE, Isaza C, Gutiérrez MI. La incidencia del síndrome de Down en Cali. Colomb Med. 1996; 27: 138-42. [ Links ]
3. García H, Salguero GA, Moreno J, Arteaga C, Giraldo A. Frecuencia de anomalías congénitas en el Instituto Materno Infantil de Bogotá. BiomEdica. 2003; 23: 161-72. [ Links ]
4. Turnpenny P, Ellard S. Elementos de genética médica. 13a ed. Barcelona; Elsevier; 2009. [ Links ]
5. Hernández D, Fisher EMC. Down syndrome genetics: unraveling a multifactorial disorder. Hum Mol Genet. 1996; 5: 1411-6. [ Links ]
6. Lamb NE, Hassold TJ. Nondisjunction. A view from the Ringside. N Engl J Med. 2004; 351: 1931-4. [ Links ]
7. Lamb NE, Yu K, Shaffer J, Feingold E, Sherman SL. Association between maternal age and meiotic recombination for trisomy 21. Am J Med Gen. 2005; 76: 91-9. [ Links ]
8. Gardiner K, Davisson M. The sequence of human chromosome 21 and implications for research into Down syndrome. Genome Biol. 2000; 1: reviews0002.1-0002.9 [ Links ]
9. Montoya JC, Satizábal JM, García-Vallejo F, Sánchez A. Perspectiva y comprensión bioquímica del síndrome de Down. El Hombre y la Máquina 2008; 30:118-29. [ Links ]
10. Toyoda A, Noguchi H, Taylor TD, Ito T, Pletcher MT, Sakaki S, et al. Comparative genomic sequence analysis of the human Chromosome 21 Down syndrome critical region. Genome Res. 2002; 12: 1323-32. [ Links ]
11. Eggermann T, Schönherr N, Spengler S, Jäger S, Denecke B, Binder G, et al. Identification of a 21q22 duplication in a Silver-Russell syndrome patient further narrows down the Down syndrome critical region. Am J Med Genet A. 2010; 152A: 356-9. [ Links ]
12. Ronan A, Fagan K, Christie L, Conroy J, Nowak NJ, Turner G. Familial 4.3 Mb duplication of 21q22 sheds new light on the Down syndrome critical region. J Med Genet. 2007; 44: 448-51. [ Links ]
13. Kollmann M, Sourjik V. In Silico biology: From simulation to understanding current biology. Curr Biol. 2007; 17: 132-4. [ Links ]
14. Bönisch C, Nieratschker SM, Orfanos NK, Hake SB. Chromatin proteomics and epigenetic regulatory circuits. Expert Rev Proteomics. 2008; 5: 105-19. [ Links ]
15. Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007; 447: 433-40. [ Links ]
16. Leeuwen FV, Steensel BV. Histone modifications: from genome-wide maps to functional insights. Genome Biol. 2005; 6: 113-19. [ Links ]
17. Ozsolak F, Song JS, Liu XS, Fisher DE. High-throughput mapping of the chromatin structure of human promoters. Nat Biotechnol. 2007; 25: 244-8. [ Links ]
18. Zhang Y, Rohde C, Tierling S, Jurkowski TP, Bock C. DNA methylation analysis of chromosome 21 gene promoters at single base pair and single allele resolution. PLoS Genet 2009; 5: e1000438-e100.1371. [ Links ]
19. Song HJ, Park J, Seo SR, Kim J, Paik SR, Chung KC. Down syndrome critical region 2 protein inhibits the transcriptional activity of peroxisome proliferator-activated receptor b in HEK293 cells. Biochem Biophys Res Commun. 2008; 376: 478-82. [ Links ]
20. Bock C, Paulsen M, Tierling S, Mikeska T, Lengauer T, Walter J. CpG Island methylation in human lymphocytes is highly correlated with DNA sequence, repeats, and predicted DNA Structure. PLoS Genet. 2006; 2: e26. [ Links ]
21. Sherman SL, Takaesu N, Freeman SB, Grantham M, Phillips C, Blackston RD. Trisomy 21: association between reduced recombination and nondisjunction. Am J Hum Genet. 1991; 49: 608-20. [ Links ]
22. Tanzi R, Watkins PC, Stewart GD, Wexler NS, Gusella JF, Haines JL. A genetic linkage map of human chromosome 21: analysis of recombination as a function of sex and age. Am J Hum Genet. 1992; 50: 551-8. [ Links ]
23. Sherman SL, Lamb NE, Feingold E. Relationship of recombination patterns and maternal age among non-disjoined chromosomes 21. Biochem Soc Trans. 2006; 34: 578-80. [ Links ]
24. Ghosh S, Feingold E, Chakraborty S, Dey SK. Telomere length is associated with types of chromosome 21 nondisjunction: a new insight into the maternal age effect on Down syndrome birth. Hum Genet. 2010; 10.1007/s00439-009-0785-8. [ Links ]
25. Saran NG, Pletcher MT, Natale JE, Cheng Y, Reeves RH. Global disruption of the cerebellar transcriptome in a Down syndrome mouse model. Hum Mol Genet. 2003; 12: 2013-9. [ Links ]

