










Services on Demand
Journal
Article
 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombia Médica
On-line version ISSN 1657-9534
Colomb. Med. vol.45 no.4 Cali Oct./Dec. 2014
Review Article
Fragile X Syndrome
Síndrome de X Frágil
1 Professor Morphology, Gynecology and Obstetrics. Hospital Universitario del Valle, University of Valle, Cali, Colombia.
2 Department of Biochemistry and Molecular Medicine; UC Davis MIND Institute, University of California, Sacramento, USA.
3 Medicine and Surgery School, University of Valle. Cali, Colombia.
4 Medical director and distinguished professor. Holder Research Chair in Fragile X syndrome. Department of Pediatrics, School of Medicine. UC Davis MIND Institute, University of California, Davis, USA.
Saldarriaga W, Tassone F, González-Teshima LY, Forero-Forero JV, Ayala-Zapata S, Hagerman R. Fragile X Syndrome. Colomb Med. 2014; 45(4): 190-8.
© 2014 Universidad del Valle. This is an Open Access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Article history
Received: 20 November 2014 - Revised: 24 November 2014 - Accepted: 1 December 2014
Corresponding author: Wilmar Saldarriaga Gil. Carrera 37 # 1oeste-45, Apto 1203B. Unidad Mirador Avalon. Cali, Colombia. Phone: +57 2 316 446 1596. E-mail: wilmar.saldarriaga@correounivalle.edu.co.
Abstract
Fragile X Syndrome (FXS) is a genetic disease due to a CGG trinucleotide expansion, named full mutation (greater than 200 CGG repeats), in the FMR1 gene locus Xq27.3; which leads to an hypermethylated region in the gene promoter therefore silencing it and lowering the expression levels of FMRP, a protein involved in synaptic plasticity and maturation. Individuals with FXS present with intellectual disability, autism, hyperactivity, long face, large or prominent ears and macroorchidism at puberty and thereafter. Most of the young children with FXS will present with language delay, sensory hyper arousal and anxiety. Girls are less affected than boys, only 25% have intellectual disability. Given the genomic features of the syndrome, there are patients with a number of triplet repeats between 55 and 200, known as premutation (PM) carriers. Most carriers have a normal IQ but some have developmental problems. The diagnosis of FXS has evolved from karyotype with special culture medium, to molecular techniques that are more sensitive and specific including PCR and Southern Blot. During the last decade, the advances in the knowledge of FXS, has led to the development of investigations on pharmaceutical management or targeted treatments for FXS. Minocycline and sertraline have shown efficacy in children.
Keyword: Fragile X Syndrome, Fragile X Mental Retardation Protein, intellectual disability, review, therapeutics, genetic counselling.
Resumen
El Síndrome de X Frágil (SXF), es una enfermedad genética debida a una expansión del trinucleótido CGG, nombrada mutación completa (más de 200 repeticiones de CGG) en el gen FMR1, locus Xq27.3; la cual lleva a una hipermetilación de la región promotora del gen, silenciándolo y disminuyendo los niveles de expresión de la proteína FMRP relacionada con la plasticidad y maduración neuronal. Los individuos con SXF presentan retardo mental, autismo, hiperactividad, cara alargada, orejas grandes o prominentes y macroorquidismo desde la pubertad. La mayoría de niños con SXF presentan retraso en el lenguaje, hiperactivación sensorial y ansiedad. Las niñas se afectan menos que los varones, solo el 25% presenta retardo mental. Dadas las características genómicas del síndrome, existen pacientes con un número de repetición de la tripleta entre 55 y 200 que se denominan portadores de la premutación. La mayoría de los portadores tienen un coeficiente intelectual normal, pero presentan problemas en el desarrollo. El diagnóstico en SXF ha evolucionado del cariotipo con medio especial de cultivo, a pruebas moleculares más sensibles y específicas incluyendo PCR y Southern blot. Durante la última década, los avances en el conocimiento sobre el SXF han permitido el desarrollo de investigaciones sobre el manejo farmacológico o tratamientos específicos para el SXF. La minociclina y la sertralina han demostrado eficacia en niños.
Palabras clave: Síndrome del Cromosoma X Frágil, Proteina retardo mental fragil X, retraso mental, revisión, terapeutica, Asesoramiento genético.
Introduction
The Fragile X Syndrome (FXS) is a genetic disease inherited through the X chromosome, which was described for the first time in 1943 by Martin and Bell1. It is actually considered the most common inherited cause of intellectual disability and the second most prevalent cause after Down syndrome. Most cases of Down syndrome are de novo but FXS is always inherited with many individuals in the family tree, either affected or a carrier of FXS. Affected men have a classic phenotype characterized by long face, large and protruding ears and macroorchidism2. The Fragile X Syndrome is caused by an abnormal expansion in the number of the trinucleotide CGG repeats located in the 5' UTR in the fragile X mental retardation 1 gene (FMR1) at Xq27.3. It is a dynamic mutation with expansion of the CGG repeat in each generation moving from the premutation range of 55 to 200 repeats and expanding to a full mutation when pass on by a women to her children3.
Patients affected with FXS have more than 200 repeats of the CGG trinucleotide. On the other hand, premutation carriers (55 to 200 repeats) although are not affected with the classic FXS phenotype, can have other medical, psychiatric and neurological problems. In the last 15 years multiple advances have been made in the description of genetic characteristics, function of the protein encoded by the FMR1 gene (FMRP), pharmacological management and the description, in carriers of the premutation, of the Fragile X associated Tremor/Ataxia Syndrome (FXTAS) and fragile X-associated primary ovarian insufficiency (FXPOI)3-5.
The objective of this review is to contribute to the dissemination of knowledge on FXS among health professionals and thus improving the diagnosis and management of these patients.
1. Epidemiology
The actual worldwide prevalence, determined by molecular assays, it's estimated in one per 5,000 men6 and in one per 4,000 to 6,000 women6,7. Ricaurte is a district of the municipality of Bolívar, located in the north of Valle del Cauca, in which there has been identified a high prevalence of mental disability, 39 intellectually disable individuals in 1124 habitants8. During the late 1990's, a study found that the cause of this disability in the region was FXS. In this study 19 patients were diagnosed with the syndrome by karyotype with G bands in folate deficient medium; furthermore clinical diagnosis was done in 16 more patients in whom the karyotype was not performed. The cases were found in 3 family names and a possible common ancestor was postulated given the migratory patterns and founding characteristics of the town9. By the year 1999 the prevalence of FXS in Ricaurte was determined as 1:38 men and 1:100 women, which exceeded 100 times the prevalence reported in literature.
A number of medical conditions and syndromes, associated with carriers of the premutation including depression, anxiety, migraine headaches, hypertension, sleep apnea, immune mediated diseases including hypothyroidism and fibromyalgia, and FXTAS and FXPOI have been described in the past 10 years10. The prevalence of the premutation in the general population is 1:130-200 women and 1:250 to 450 men11,12. Tremor/Ataxia Syndrome occurs in approximately 40% of men with the premutation and 16% of women, whereas FXPOI occurs in approximately 16 to 20% of women with the premutation2,10.
2. Genomics
The Fragile X Syndrome is caused by an alteration in the FMR1 gene, with locus Xq27.3. This gene harbors a CGG repeat within the 5' Untranslated Region. Depending on the number of repetitions, 4 types of alleles are defined with different clinical manifestations3,13: Normal alleles, up to 44 CGG repeats; premutation (PM) alleles, between 55 and 200 and full mutation alleles (FM) with more than 200 repeats. The fourth type of allele is named "grey zone" or intermediate allele and contains between 45 and 54 repeats and it has been proposed as a precursor for PM alleles.
The silencing of the FMR1 gene is the result of a series of complex epigenetic modifications following the expansion of the trinucleotide repeat14. The FM alleles undergo a methylation process in the CpG island within the gene promoter and in the CGG repeats15,16. In male patients with the FM allele, every cytosine within the CpG island is methylated, contrarily to healthy individuals who lack of any methylation17. A recently discovered boundary sequence in the FMR1 gene found 650 to 800 nucleotides upstream from the repeated region that suffers methylation. This boundary sequence, because of its chromatin interaction, limits the hypermethylated region of the genome, protecting the promoter of FMR1 from possible methylation15,16,18. In individuals with FXS the methylated boundary sequence is lost allowing the expansion of the methylation up to the promoter of the FMR1 gene. This findings strongly suggest that changes in the sequence of nucleotides and conformational structure of the chromatin of the boundary sequences would favor the epigenetic changes that would induce FMR1 silencing and ultimately preventing FMRP production19.
FXS is usually caused by the methylation and gene silencing associated with the full mutation although deletions of the coding region of the gene can also lead to absence of FMRP. In addition, point mutations or reading frame shifts can also occur leading to a functional deficit of the protein FMRP and the consequent phenotype; however, these genomic changes account only for less than 1% of all FXS cases described20.
The clinical involvement in those with a premutation is caused by a different mechanism than FXS, which is elevated FMR1 mRNA levels leading to RNA toxicity. 2-8 folds normal levels of mRNA are observed in premutation carriers and the excess such mRNA lead to the sequestration of important proteins for neuronal function by the hairpin structures that form in the CGG repeats. RNA toxicity causes the neurons to die earlier in culture and so the carriers are at risk for late onset disorders including FXTAS and FXPOI10.
The FMRP is a RNA binding protein that shows preference towards RNA homopolymers21 as for certain subgroups of cerebral transcripts22. The protein encoded by the FMR1 gene is involved in the regulation of the RNA stability, subcellular transport and translation of neural mRNAs that codify for proteins involved in the synapsis development, neural plasticity and brain development23-25. Various studies have revealed that in the absence of this protein, a wide range of neural mRNAs are altered, augmenting neural protein synthesis and resulting in dendritic spine dysmorphogenesis and an excitation/inhibition imbalance (Glutamate/GABA), phenomena present in FXS26,27. The dendritic spine dysmorphogenesis plays a role in the clinical manifestations of the syndrome, due to the weak synaptic connections leading to intellectual deficits and behavioral problems. Multiple neurotransmitter systems are impaired because of the lack of FMRP and there is enhanced protein production in the hippocampus and throughout the brain3.
3. Heritability
FXS heritability does not have a Mendelian classic inheritance pattern. It depends on the number of trinucleotide CGG repeats within the promoter of the FMR1 gene28. The transition from PM to FM alleles occurs because of expansion phenomena during the transmission of the maternal X chromosome carrying PM, to her children2. This expansion does not occur during the transmission of the paternal X chromosome, with the PM, to their daughters 28. So all daughters of men with the premutation will be obligate carriers of the premutation but then these daughters have a 50% risk to have children with FXS.
3.1. Dynamic of the mutation
The risk of transition from PM to FM in the descendants depends on the number of trinucleotide repeats in the PM allele, reaching ∼100% for PM alleles with more than 99 repeats29,30. The expansion from PM to FM in meiosis can occur in alleles with as little as 56 CGG repeats31; the odds that this occurs depends on the range of repeats in which the patient is classified, number of AGG interruptions and age of the mother (Table 1)29,30. Normally there is an AGG anchor with every 9 or 10 CGG repeats in FMR1. The anchors can modify the risk for expansion of the CGG repeat when passed on by the mother. Women with the premutation and 2 AGG anchors have a lower risk of expansion to the full mutation compared to women with no AGG anchors29,30.
The intermediate or grey zone alleles are those that possess between 45 and 54 repeats and are proposed as precursors of PM alleles. The transition from grey zone alleles to PM alleles occur because of paternal or maternal meiosis instability32.
3.2. Inheritance and recurrence risk
Men. The majority of the men with FM usually do not reproduce with only 1% of them reported to have offspring2. Male patients with FM and FXS have a 100% chance to pass on the premutation to their daughters so their daughters will only be carriers and usually they do not present with intellectual disabilities. There is loss of the FM in the formation of the sperm and only the premutation is passed on. All of their sons will receive the Y chromosome so they will not be affected with the FMR1 mutation. Meanwhile, men with the PM will transmitt only the PM to their daughters and the number of repeats is relatively stable2,31.
Women. It's noteworthy that carriers of the PM can expand their allele to FM with odds depending on the number of repeats, number of AGG interruptions and age (Table 1). The impact of the AGG interruptions is attributed to the decrease of DNA polymerase slippage in replication. Therefore, this interruptions give stability in the gene's transmission but does not affect gene's transcription nor translation29,30.
Besides, according to the newborn's sex, the clinical characteristics are going to differ. Male patients with FM will develop mental impairment. On the contrary, daughters who inherit the FM have a 30% chance to have a normal intelligence quotient, 25% to have intellectual disability with IQ less than 70, nonetheless they can present learning deficit (60%) and emotional difficulties (70%)2.
4. Fragile X Syndrome clinical characteristics
The FXS cognitive and emotional phenotype will depend as well on the amount of FMRP that is produced, which depends on the number of repeats and the methylation degree of the FMR1. Depending on the FMRP concentration, a clinical spectrum develops. When FMRP levels are not very low the symptoms are less severe, there's a moderate emotional and learning difficulties and a normal intelligence quotient. If the production of the FMRP protein decreases or stops, a severe cognitive deficit develops causing mental impairment2.
Particular physical characteristics also depend and the production of FMRP. Thus, the 80% of the patients with FXS will have one or more of their common facial characteristics (Table 2 and Fig. 1)2. The diagnosis is suspected in men with mental impairment, particular facial characteristics as long face, large and protruded ears, and macroorchidism; this phenotype allows the distinction of patients with FXS after puberty to the patients with mental impairment and not associated with FXS2,20. However all individuals with intellectual disability or autism should have the fragile X DNA test because sometimes the physical features are not obvious or not present, particularly in young children.
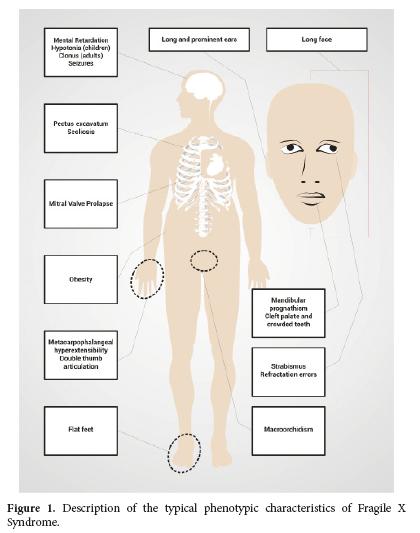
Women with FM and FXS are going to present a wider range of phenotypic characteristics than men, depending on the activation ratio of the affected X chromosome AR in peripheral blood (AR= percentage or ratio of cells with the normal allele present on active X chromosome, so that higher AR correspond to higher FMRP levels produced by the normal FMR1 allele). The severity of the phenotype in women can vary from typical physical characteristics (Table 2 and Fig. 1) and mental impairment to the absence of physical phenotype together with a mild learning disability. Seventy per cent of the women with the FM present some degree of cognitive impairment2.
Premutation carriers usually have a normal IQ with mild or no physical features; however, approximately 20% of women develop FXPOI or FXTAS, although 40% of male carriers develop FXTAS. Tremor/Ataxia Syndrome is a late onset progressive neurodegenerative disorder10, characterized by neurological deficits that include progressive intention tremor, cerebellar ataxia, parkinsonism, neuropathy and autonomic dysfunction33-35. Fragile X-associated primary ovarian insufficiency, which indicate the cessation of the menses before 40 years old, affects women by producing a series of complications in their fertility and reproduction, because it causes menstrual cycle irregularities, infertility and ovary hormonal deficiency36.
5. Diagnosis
Initially, the diagnosis of FXS was done through karyotype, which allowed the observation of the distal narrowing of the long arm of the X chromosome in the band 27.3 (Xq27.3-23.8) using the light microscope. The findings of distal constrictions can be done in different chromosomes, and are known as fragile sites, from where FXS is named2.
Nowadays there are several molecular tests available for the diagnosis of FXS which are far more sensitive and specific than the karyotype11. Besides allowing the diagnosis of patients with the FM and FXS, theses tests allow the identification of carriers of the PM, which are individuals typically with a normal IQ, but the female carriers have a high risk of having children with FXS. As well there are molecular tests that allow the quantification of messenger RNA (mRNA) and of FMRP protein, allowing a better understanding of the physiopathology of the disease by correlating the results to the phenotype of the FM and PM patients2. Polymerase chain reaction (PCR) and Southern blot are the routine tests for the DNA diagnosis of FXS which allow determining the number of CGG repeats and the methylation status of the FMR1 gene.
The PCR through the use of specific primers for the FMR1 gene allows the amplification of the region that contains the CGG repeat and, can identify patients with an expanded FMR1 allele particularly in the premutation but also in the full mutation range37.Usually Southern blot analysis is utilized to better characterize alleles in the full mutation range and to determine the methylation status38. However, with the new PCR techniques that use double primers for a nested PCR, the quantification of CGG triplet repetition and identification of FM and PM patients is possible37,39.
The tests for the diagnosis of FXS must be ordered for patients with intellectual disability and/ or autism and additionally any of the following features: distinct facial features as large protruded ears, long face, among others; family history of intellectual disability, autism, macroorchidism (Fig. 2)
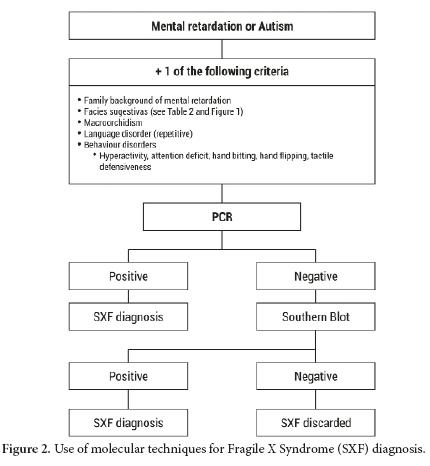
Once it is determined that the patient has the FM or PM for FXS, DNA molecular testing must be done on all the family members that are suspected through the analysis of the pedigree of being carriers. Likewise family members with tremor, ataxia, neurological symptoms or early ovarian insufficiency would also be candidates for the molecular tests40.
The diagnosis of FXS can be done even in fetus of pregnant carriers or patients with FXS using the same molecular tests mentioned above (Southern blot and PCR) on chorionic villus sampling40.
6. Differential diagnosis
The differential diagnosis includes Sotos syndrome, Prader-Willi and Klinefelter as they share particular characteristics. For each one of these syndromes there are specific molecular tests that help to confirm the diagnosis. These tests will be ordered according to the phenotypic findings and clinical analysis of the patients (Table 3)2,41,42. The most frequent clinical findings among these syndromes that can be contrasted with FXS are:
- Sotos syndrome: intellectual disabilities, tall stature, macrocephaly, and epilepsy.
- Prader-Willi syndrome: intellectual disabilities, obesity, short stature, and hypogenitalism. A subgroup of patients with FXS will have a Prader-Willi phenotype but will not have a deletion at 15 q11-13 region, although the level of CYFIP protein from this region is low.
- Klinefelter syndrome: tall stature, hypogenitalism, intellectual disabilities (20%)
- FRAXE: intellectual disabilities, language impairment, hyperactivity, autistic behavior (due to the abnormal repetition of CCG triplet in the FMR2 gene)
- intellectual disabilities and chromosome fragility in other fragile sites have been described (FRAXD and FRAXF genes).
Other less frequent syndromes, with prevalence lower than one in 50,000, have similar genetic and physiopathogenic mechanisms as well as phenotype to FXS. Among these syndromes there are Fragile X syndrome E and Fragile X syndrome F, due to the alteration of the FMR2, FAM11A and FRAXD genes respectively. If the phenotype of the patient highly suggests FXS and the Southern blot results come out negative, molecular testing for genes mentioned above should be considered.
7. Treatment
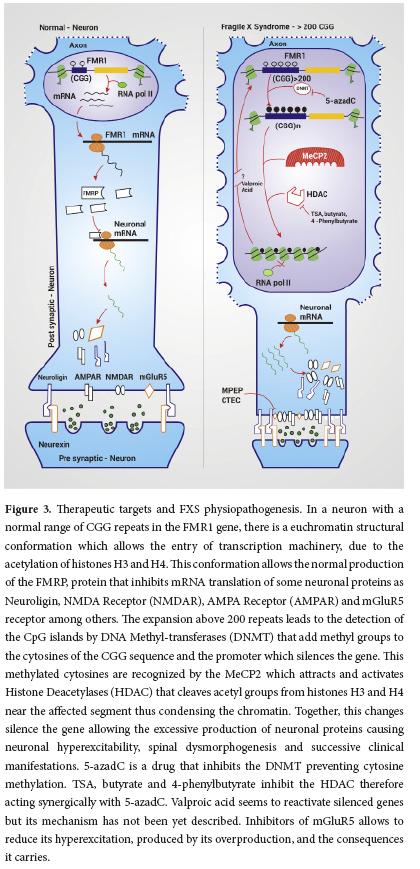
Multiple studies have been carried out in the attempt to develop target treatments for FXS that can improve some of the symptoms and the life quality of the affected patient. These investigations have had different approaches according to their objectives, whether it is the activation of the FMR1 gene or the treatment of the symptoms associated to this disease. The mechanism of action of some of the medications used focus on epigenetic modulation, glutamatergic system and regulation of the translation of FMRP target mRNAs (Fig. 3.)
Even though folic acid decreased the observation of fragile sites in the X chromosome in in vitro models of FXS, this drug doesn't have the same effect in vivo. Its benefits could derive from the role of this micronutrient in the methylation and hydroxylation of neurotransmitters, reactions implicated in their synthesis and metabolism2. The first clinical trials with this supplement in the 1980´s reported that folic acid gave a similar positive effect to the one obtained with the use of stimulants, specifically in motor coordination, language and speech. However, the effectiveness of this drug has been difficult to demonstrate in controlled studies, limiting its potential as a possible treatment and contraindicating it for patients with seizures and FXS2,43.
Valproic acid, known as a silent gene reactivator3,44, is a weak reactivator of the FM alleles in FXS3,45. It has been reported that the administration of this drug to patients with FXS improves their attention deficit hyperactivity disorder (ADHD). In patients with generalized tonic-clonic seizures, absence crisis and partial seizures this is the drug of choice; as by increasing the levels of GABA and decreasing dopamine it is an excellent mood stabilizer and can be used in cases of bipolar disorder in adults and even in children2,3,46.
Likewise, L - Acetyl - carnitine also seems to inhibit the formation of fragile sites in the X chromosome. Its administration to patients with FXS improves significantly their attention deficit hyperactivity disorder, however it has no effect in the methylation of the FMR1 gene, nor its expression2,3. The use of this drug in the treatment of FXS patients could be indicated; however the reported studies are not conclusive.
The best medications available for treatment of ADHD symptoms in FXS are the stimulants including preparations of methylphenidate or mixed amphetamine salts. However, these medications should not be used in children under age five because they can often cause irritability47. Nevertheless, if severe ADHD symptoms occur under age five then guanfacine or clonidine can be helpful. Clonidine can also be used to treat the sleep disturbance that is a common problem for young children with FXS. Alternatively, melatonin at a dose of 1 to 3 mg at bedtime works well for facilitating sleep in children with FXS48.
The use of mGluR antagonists as 2-methyl-6-phenylethynyl-pyridine and CTEC, has given as a result the recovery of the dendritic spines morphology, protein synthesis, hippocampus atrophy and partially the macroorchidism in animal models3,49. However, the use of mGluR5 antagonists have not demonstrated efficacy in adolescents and adults with FXS in controlled trials2.
One of the great interests and pharmacologic targets for the treatment of FXS is the restoration of the FMR1 gene activity by decreasing the DNA methylation and altering the H3 and H4 histones acetylation code. The use of 5-azadC (methyltransferase inhibitor) along with histone deacetylase inhibitors like TSA, butyrate and 4-phenylbutyrate have been proven to act sinergically for this purpose. Both drugs have been tested solely in vitro, due to the risk of inducing cellular apoptosis in vivo3.
On the other hand, due the absence of FMRP protein in FXS, which regulate synaptic stimulation50, an overproduction of metalloproteinase 9 (MMP-9) is produced in response to the normal synaptic stimulations in this disease51, which has been associated with several pathologic conditions, particularly seizures and cerebrovascular accidents52. Active MMP-9 regulates the pericellular environment through the cleavage of protein components. This MMP-9 function has been associated with synaptic changes associated with learning and memory53. Treatment with minocycline for three months in patients with FXS has been proven, in some cases, to decrease MMP-9 activity independent of age and doses54. A controlled trial of minocycline (doses 25 to 100 mg per day) in children ages 3.5 to 16 years demonstrated efficacy in behavioral improvements and with mood and anxiety problems55
Minocycline can be used clinically in treatment of children and adults with FXS but when used in children under 8 it can lead to darkening of the teeth. However, when used in young children it may have its greatest effect in strengthening synaptic connections and enhancing cognitive development. The physician should discuss the side effects of minocycline with the family before prescribing Minocycline, which can also lead to loose stools usually improving with a probiotic. The darkening of the teeth can be fixed cosmetically when the child is older. Rarely minocycline can cause elevation of the antinuclear antibody and if a rash or swollen joints, visual problems or severe headache occur then minocycline should be discontinued55.
Due to the high frequency of mood, anxiety and behavior disorders in patients with this syndrome and in carriers of the PM, serotonin selective reuptake inhibitors have been used to treat effectively aggressive behavior, anxiety, depression among others2. The use of sertraline in FXS is a subject of current investigations; articles have reported its positive effect in language development in young children (2 to 6 years) with FXS among other virtues56. However, in about 20% of cases an SSRI can worsen aggression because of hyperarousal especially if the dose is high. Therefore, if aggression develops with the use of an SSRI, the dose should be lowered or discontinued and a mood stabilizer, such as aripiprazole or risperidone should be started. Lithium is also an excellent mood stabilizer in FXS and a targeted treatment for FXS47.
Besides all of the pharmacologic options mentioned above, several authors, remark on the necessity of developing cognitive and behavior therapies, as well as educational and behavioral intervention for these patients, to be able to strengthen their social abilities, reading abilities, adaptive behavior and support network57-61. Children with FXS are also typically very good with the computer so applications that focus on language, reading, math, or social deficits can be used with the help of the teacher, therapist or parent.
8. Conclusion
In Colombia the prevalence of FXS hasn´t been reported; however there are many patients with the full mutation and many premutation carriers with an endemic focus in Ricaurte, Valle del Cauca. The knowledge of the genomics, physiopathology of the disease and the function of the FMRP in the central nervous system, the heritability and phenotypical findings in the carriers have increased in the last decade. This has allowed the stimulation of multiple investigations, especially in pharmacologic interventions with targeted treatments that can reverse the neurobiology of FXS. However, the drugs that increase the FMRP levels or the expression of the FMR1 gene haven´t been used in vivo due to safety reasons; other drugs as valproic acid, sertraline, minocycline among others, have given good results in specific symptoms of FXS patients, although they should be used according to each individual response.
Conflict of interest: All authors do not have any possible conflicts of interest.
References
1. Martin JP, Bell J. A pedigree of mental defect showing sex-linkage. J Neurol Psychiatry. 1943; 6(3-4): 154-7. [ Links ]
2. Hagerman RJ, Hagerman PJ. Fragile X Syndrome. 3. Baltimore: The John Hopkins University Press; 2002. [ Links ]
3. Bagni C, Tassone F, Neri G, Hagerman R. Fragile X syndrome: Causes, diagnosis, mechanisms, and therapeutics. J Clin Invest. 2012; 122(12): 4314-22. [ Links ]
4. Liu Y, Winarni T, Zhang L, Tassone F, Hagerman R. Fragile X-associated tremor/ataxia syndrome (FXTAS) in grey zone carriers. Clin Genet. 2013; 84: 74-7. [ Links ]
5. Hall D, Tassone F, Klepitskaya O, Leehey M. Fragile X associated tremor ataxia syndrome in FMR1 gray zone allele carriers. Mov Disord. 2012; 27(2): 296-300. [ Links ]
6. Coffee B, Keith K, Albizua I, Malone T, Mowrey J, Sherman SL, et al. Incidence of Fragile X syndrome by newborn screening for methylated FMR1 DNA. Am J Hum Genet. 2009; 85: 503-14. [ Links ]
7. Hirst MC, Knight SJ, Christodoulou Z, Grewal PK, Fryns JP, Davies KE. Origins of the fragile X syndrome mutation. J Med Genet. 1993; 30(8): 647-50. [ Links ]
8. Gardeazabal AG. El divino. Bogota: Plaza Janes; 1986. [ Links ]
9. Payan C, Saldarriaga W, Isaza C, Alzate A. Estudio foco endémico de retardo mental en Ricaurte Valle. Acta Biológica Colombiana. 2001; 6(2): 88. [ Links ]
10. Hagerman R, Hagerman P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 2013; 12(8): 786-98. [ Links ]
11. Fernandez-Carvajal I, Walichiewicz P, Xiaosen X, Pan R, Hagerman PJ, Tassone F. Screening for expanded alleles of the FMR1 gene in blood spots from newborn males in a Spanish population. J Mol Diagn. 2009; 11(4): 324-9. [ Links ]
12. Tassone F, Iong KP, Tong TH, Lo J, Gane LW, Berry-Kravis E, et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 2012; 4(12): 1-113. [ Links ]
13. Center for Disease Control and Prevention. Fragile X syndrome (FXS). Accessed 20 November 2014. Available from: http://www.cdc.gov/ncbddd/fxs/index.html. [ Links ]
14. Pietrobono R, Tabolacci E, Zalfa F, Zito I, Terracciano A, Moscato U, et al. Molecular dissection of the events leading to inactivation of the FMR1 gene. Hum Mol Genet. 2005; 14(2): 267-77. [ Links ]
15. Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, et al. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991; 66(4): 817-22. [ Links ]
16. Naumann A, Hochstein N, Weber S, Fanning E, Doerfler W. A distinct DNA-methylation boundary in the 5'-upstream sequence of the FMR1 promoter binds nuclear proteins and is lost in fragile X syndrome. Am J Hum Genet. 2009; 85(5): 606-16. [ Links ]
17. Stöger R, Kajimura TM, Brown WT, Laird CD. Epigenetic variation illustrated by DNA methylation patterns of the fragile-X gene FMR1. Hum Mol Genet. 1997; 6(11): 1791-801. [ Links ]
18. Malter HE, Iber JC, Willemsen R, de Graaff E, Tarleton JC, Leisti J, et al. Characterization of the full fragile X syndrome mutation in fetal gametes. Nat Genet. 1997; 15(2): 165-9. [ Links ]
19. Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, et al. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992; 1(6): 397-400. [ Links ]
20. Kniffin CL, Jackson JF. Fragile x mental retardation syndrome. Accessed: 20 November 2014. Available from: http://omim.org/clinicalSynopsis/300624?search=fragilexhighlight=x fragile. [ Links ]
21. Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell. 1993; 74(2): 291-8. [ Links ]
22. Ashley CT, Wilkinson KD, Reines D, Warren ST. FMR1 protein: conserved RNP family domains and selective RNA binding. Science. 1993; 262(5133): 563-6. [ Links ]
23. Darnell JC, Van Driesche SJ, Zhang C, Hung KYS, Mele A, Fraser CE, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247-61. [ Links ]
24. Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, et al. Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell. 2001; 107(4): 477-87. [ Links ]
25. Chen L, Yun SW, Seto J, Liu W, Toth M. The fragile X mental retardation protein binds and regulates a novel class of mRNAs containing U rich target sequences. Neuroscience. 2003; 120(4): 1005-17. [ Links ]
26. Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004; 27(7): 370-7. [ Links ]
27. Gatto CL, Broadie K. Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front Synaptic Neurosci. 2010; 2: 4. [ Links ]
28. Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991; 65(5): 905-14. [ Links ]
29. Yrigollen CM, Martorell L, Durbin-Johnson B, Naudo M, Genoves J, Murgia A, et al. AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J Neurodev Disord. 2014; 6(1): 1-11. [ Links ]
30. Yrigollen CM, Durbin-Johnson B, Gane L, Nelson DL, Hagerman R, Hagerman PJ, et al. AGG interruptions within the maternal FMR1 gene reduce the risk of offspring with fragile X syndrome. Genetics Med. 2012; 14(8): 729-36. [ Links ]
31. Fernandez-Carvajal I, Lopez Posadas B, Pan R, Raske C, Hagerman PJ, Tassone F. Expansion of an FMR1 grey-zone allele to a full mutation in two generations. J Mol Diagn. 2009; 11(4): 306-10. [ Links ]
32. Nolin SL, Brown WT, Glicksman A, Houck GE, Gargano AD, Sullivan A, et al. Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am J Hum Genet. 2003; 72(2): 454-64. [ Links ]
33. Berry-Kravis E, Abrams L, Coffey SM, Hall DA, Greco C, Gane LW, et al. Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov Disord. 2007; 22(14): 2018-2030. [ Links ]
34. Seltzer MM, Baker MW, Hong J, Maenner M, Greenberg J, Mandel D. Prevalence of CGG expansions of the FMR1 gene in a US population-based sample. Am J Med Genet B Neuropsychiatr Genet. 2012; 159B(5): 589-97. [ Links ]
35. Bourgeois JA, Cogswell JB, Hessl D, Zhang L, Ono MY, Tassone F, et al. Cognitive, anxiety and mood disorders in the fragile X-associated tremor/ataxia syndrome. Gen Hosp Psychiatry. 2007; 29(4): 349-56. [ Links ]
36. Martin JR, Arici A. Fragile X and reproduction. Curr Opin Obstet Gynecol. 2008; 20(3): 216-20. [ Links ]
37. Filipovic-Sadic S, Sah S, Chen L, Krosting J, Sekinger E, Zhang W, et al. A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin Chem. 2010; 56: 399-408. [ Links ]
38. Tassone F, Pan R, Amiri K, Taylor AK, Hagerman PJ. A rapid polymerase chain reaction based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J Mol Diagn. 2008; 10: 43-9. [ Links ]
39. Chen L, Hadd A, Sah S, Filipovic-Sadic S, Krosting J, Sekinger E, et al. An information-rich CGG repeat primed PCR that detects the full range of fragile X expanded alleles and minimizes the need for southern blot analysis. J Mol Diagn. 2010; 12: 589-600. [ Links ]
40. Firth HV, Hurst JA, Hall JG. Oxford Desk Reference? Oxford: Oxford University Press; 2005. [ Links ]
41. O'Neil MJF, Jackson JF. SOTOS SYNDROME 1 SOTOS1. Accessed: 20 November 2014. Available from: http://omim.org/clinicalSynopsis/117550?search=highlight=sotos. [ Links ]
42. Hamosh A, Jackson JF. PRADER-WILLI SYNDROME PWS. Accessed: 20 November 2014. Available from: http://omim.org/clinicalSynopsis/176270?search=praderhighlight=prader. [ Links ]
43. Rueda JR, Ballesteros J, Guillen V, Tejada MI, Solà I. Folic acid for fragile X syndrome. Cochrane database Syst Rev. 2011;(5):CD008476. [ Links ]
44. Detich N, Bovenzi V, Szyf M. Valproate induces replication-independent active DNA demethylation. J Biol Chem. 2003; 278(30): 27586-92. [ Links ]
45. Tabolacci E, De Pascalis I, Accadia M, Terracciano A, Moscato U, Chiurazzi P, et al. Modest reactivation of the mutant FMR1 gene by valproic acid is accompanied by histone modifications but not DNA demethylation. Pharmacogenet Genomics. 2008; 18(8): 738-41. [ Links ]
46. Torrioli M, Vernacotola S, Setini C, Bevilacqua F, Martinelli D, Snape M, et al. Treatment with valproic acid ameliorates ADHD symptoms in fragile X syndrome boys. Am J Med Genet A. 2010; 152A(6): 1420-7. [ Links ]
47. Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, et al. Advances in the treatment of fragile X syndrome. Pediatrics. 2009; 123(1): 378-90. [ Links ]
48. Wirojanan J, Jacquemont S, Diaz R, Bacalman S, Anders TF, Hagerman RJ, et al. The efficacy of melatonin for sleep problems in children with autism, fragile X syndrome, or autism and fragile X syndrome. J Clin Sleep Med. 2009; 5: 145-50. [ Links ]
49. Su T, Fan H-X, Jiang T, Sun W-W, Den W-Y, Gao M-M, et al. Early continuous inhibition of group 1 mGlu signaling partially rescues dendritic spine abnormalities in the Fmr1 knockout mouse model for fragile X syndrome. Psychopharmacology. 2011; 215(2): 291-300. [ Links ]
50. Dziembowska M, Pretto DI, Janusz A, Kaczmarek L, Leigh MJ, Gabriel N, et al. High MMP-9 activity levels in fragile X syndrome are lowered by minocycline. Am J Med Genet A. 2013; 161A(8): 1897-1903. [ Links ]
51. Bilousova T V, Dansie L, Ngo M, Aye J, Charles JR, Ethell DW, et al. Minocycline promotes dendritic spine maturation and improves behavioural performance in the fragile X mouse model. J Med Genet. 2009; 46(2): 94-102. [ Links ]
52. Wilczynski GM, Konopacki FA, Wilczek E, Lasiecka Z, Gorlewicz A, Michaluk P, et al. Important role of matrix metalloproteinase 9 in epileptogenesis. J Cell Biol. 2008; 180(5): 1021-35. [ Links ]
53. Wang X, Bozdagi O, Nikitczuk JS, Zhai ZW, Zhou Q, Huntley GW. Extracellular proteolysis by matrix metalloproteinase-9 drives dendritic spine enlargement and long-term potentiation coordinately. Proc Natl Acad Sci U S A. 2008; 105(49): 19520-5. [ Links ]
54. Dziembowska M, Milek J, Janusz A, Rejmak E, Romanowska E, Gorkiewicz T, et al. Activity-dependent local translation of matrix metalloproteinase-9. J Neurosci. 2012; 32(42): 14538-47. [ Links ]
55. Leigh MJS, Nguyen D V, Mu Y, Winarni TI, Schneider A, Chechi T, et al. A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile x syndrome. J Dev Behav Pediatr. 2013; 34(3): 147-55. [ Links ]
56. Indah Winarni T, Chonchaiya W, Adams E, Au J, Mu Y, Rivera SM, et al. Sertraline may improve language developmental trajectory in young children with fragile X syndrome: A retrospective chart review. Autism Res Treat. 2012; 2012: 104317. [ Links ]
57. Moskowitz LJ, Carr EG, Durand VM. Behavioral intervention for problem behavior in children with fragile X syndrome. Am J Intellect Dev Disabil. 2011; 116(6): 457-8. [ Links ]
58. Turk J. Fragile X syndrome: lifespan developmental implications for those without as well as with intellectual disability. Curr Opin Psychiatry. 2011; 24(5): 387-97. [ Links ]
59. Dew-Hughes D. Educating children with fragile X syndrome. Florence: Routledge; 2003. [ Links ]
60. Dyer-Friedman J, Glaser B, Hessl D, Johnston C, Huffman LC, Taylor A, et al. Genetic and environmental influences on the cognitive outcomes of children with fragile X syndrome. J Am Acad Child Adolesc Psychiatry. 2002; 41(3): 237-44. [ Links ]
61. Hessl D, Dyer-Friedman J, Glaser B, Wisbeck J, Barajas RG, Taylor A, et al. The influence of environmental and genetic factors on behavior problems and autistic symptoms in boys and girls with fragile X syndrome. Pediatrics. 2001; 108(5): E88. [ Links ]
