










Serviços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkColombia Médica
versão On-line ISSN 1657-9534
Colomb. Med. vol.47 no.3 Cali set. 2016
Articles
Hiperplasia suprarrenal congénita por déficit de 11-beta-hidroxilasa: descripción de una nueva mutación, R384X
1 Universidad del Valle, Cali, Colombia
2 Hospital Universitario del Valle, Cali, Colombia
3 Instituto Cisalva, Universidad del Valle, Cali, Colombia
4 Fundación SCISCO, Cali, Colombia
5 Fundación Clínica Valle del Lili, Cali, Colombia
6 Fundación Clínica Infantil Club Noel, Cali, Colombia
Descripción del Caso:
Se presenta el fenotipo de una nueva mutación heterocigota compuesta en los genes Q356X y R384X que codifican la enzima 11-beta-hidroxilada
Tratamiento y Resultados:
Manejo con terapia de reemplazo hormonal con corticoide y antihipertensivo con beta-bloqueador con lo que se logró controlar los cambios físicos y los niveles de tensión arterial.
Relevancia Clínica:
Según las características fenotípicas del paciente se infiere que la mutación R384X acarrea una carga adicional a la mutación Q356X, esta última descrita como causa de deficiencia de 11-beta-hidroxilasa. La descripción de nuevos genotipos, como en este caso, permite ampliar la comprensión de la carga hereditaria y descifrar los diversos factores que llevan a que esta patología, así como las demás formas de hiperplasia suprarrenal congénita (HSC), se presenten con un amplio espectro de cuadros clínicos. Esto permite resaltar la importancia de una descripción completa del perfil genético del paciente con HSC y de sus padres.
Palabras clave: Hiperplasia suprarrenal congénita; hiperplasia suprarrenal; mutación; hormona adrenocorticotropica, virilismo, 11-beta-hidroxilasa
Case Description:
It is presented the phenotype of a new compound heterozygous mutation of the genes R384X and Q356X encoding the enzyme of 11-beta-hydroxylase
Treatment and Outcome:
Managed with hormone replacement therapy (corticosteroid) and antihypertensive therapy (beta-blocker), resulting in the control of physical changes and levels of arterial tension.
Clinical Relevance:
According to the phenotypic characteristics of the patient, it is inferred that the R384X mutation carries an additional burden on the Q356X mutation, with the latter previously described as a cause of 11-beta-hydroxylase deficiency. The description of a new genotype, as in this case, expands the understanding of the hereditary burden and deciphers the various factors that lead to this pathology as well as the other forms of congenital adrenal hyperplasia (CAH), presenting with a broad spectrum of clinical presentations. This study highlights the importance of a complete description of the patient's CAH genetic profile as well as their parents' genetic profile.
Keywords: Adrenal hyperplasia congenital; hyperplasia adrenal glands; mutation; adrenocorticotropic hormone, Virilism, 11-beta-hydroxylase deficiency
Introducción
La hiperplasia adrenal congénita (HAC) es un grupo de desórdenes enzimáticos en uno de los pasos de la producción de cortisol 1. La segunda causa más frecuente de HAC es el déficit de 11-beta-hidroxilasa (6-8%) 2, con una prevalencia de 1 por 100.000 habitantes 3, y descrita principalmente en población Judía Marroquí 4. Clínicamente se caracteriza por una virilización acompañada de hipertensión arterial secundaria a retención de sodio y aumento de volumen 1.
Tras el aislamiento del DNA codificante para la 11 beta-hidroxilasa en el cromosoma 8q21, se encontraron un par de genes, el CYP11B1 asociado a la expresión de 11 beta-hidroxilasa, y el CYP11B2, asociado a la expresión de aldosterona sintasa 5. Enzimas estrechamente relacionadas por su relevancia en la síntesis de corticosterona y aldosterona, respectivamente. Desde entonces se han descrito diversas mutaciones asociadas al gen CYP11B1 asociadas al déficit de 11-beta-hidroxilasa, la gran mayoría con una distribución geográfica o demográfica distinguida, entre estas; la mutación p.R448H descrita en judíos inmigrantes desde marruecos 4, mutación de g.4671_4672insC y g.2791G>A descritas en Brasil 6, las mutaciones p.Q356X (c.1066C>T, rs146124466) y p.G379V (c.1136G>T) descritas en Túnez y originarios africanos 3,7, y más recientemente las mutaciones p.E67fs y p.R448H descritas en familias croatas 8.
A continuación se describe un caso clínico de hiperplasia adrenal congénita por déficit de 11-beta-hidroxilasa en donde se encontró una mutación genética no descrita previamente en la literatura.
Caso clínico
Paciente de 2 años y 9 meses de edad, raza afro descendiente, con sexo asignado masculino, él y su familia, originarios y procedentes de Buga, Valle del Cauca, Colombia, quien consultó por primera vez al servicio de Endocrinología Pediátrica en mayo de 2012 por aparición de vello púbico, acné, olor axilar, aumento de tamaño del pene y talla alta. Dentro de sus antecedentes se encontró que fue un recién nacido a término, con peso adecuado, producto del segundo embarazo de madre con gravidez dos-partos dos, quien realizó los controles prenatales y no presentó ninguna complicación en el curso del embarazo, cuyos únicos antecedentes de relevancia fueron la presencia de hernia umbilical no operada e historia de cuadros respiratorios previos y exposición al humo de cigarrillo. No se reportó exposición a tóxicos, ni antecedentes familiares de relevancia.
Hallazgos clínicos
Se trató de un paciente que aparentaba mayor edad cronológica, con cifras tensionales por encima del percentil 99, peso y talla por encima de 2 desviaciones estándar (DE) para la edad, presencia de macro y microcomedones en frente y mejillas, crecimiento de pene, testículos no palpables en escroto o canal inguinal, hipospadias del glande e hiperpigmentación de genitales.
Evaluación diagnóstica
Ante los hallazgos clínicos del paciente se consideró como mayor sospecha diagnostica la hiperplasia adrenal congénita (HAC) y se solicitaron los exámenes de laboratorio descritos en la Tabla 1. En los estudios imagenológicos y complementarios, se evidenciaron ambas suprarrenales normales, presencia de útero y ausencia de genitales, edad ósea de 10 años y cariotipo 46XX. No se toman niveles de hormona adrenocorticotropa (ACTH) por problemas administrativos, sin embargo los hallazgos reportados se consideraron suficientes para corroborar el diagnostico de HAC por déficit de 11-beta-hidroxilasa con compromiso de la diferenciación sexual por virilización Prader V.
Tabla 1. Exámenes de Laboratorio: Paciente con Hiperplasia Suprarenal Congénita por Déficit de 11-Betahidroxilasa
| Examen | Resultado |
|---|---|
| 17 OH-progesterona (ng/dL) | 5,200 |
| Deshidroepiandrosterona -Sulfato (mcg/dL) | 61.5 |
| Aldosterona (100 pg/dL) | |
| Renina (pg/dL) | <1.6 |
| Cortisol (nmol/L) | 21.5 |
| Testosterona (mg/dL) | 1.66 |
| Sodio (mEq/L) | 141 |
| Potasio (mEq/L) | 3.7 |
| Cloro (mEq/L) | 101 |
| Desoxicorticosterona | Elevada |
| Androstenediona | elevada |
Se realizó un análisis por secuenciación automática de los exones y regiones intrónicas flanqueantes del gen CYP11B1 a partir de ADN genómico del paciente y de la madre después de consentimiento informado. Se encontró una mutación Q356X en estado heterocigota a nivel del exón 6 ya descrita en pacientes con deficiencia de 11-beta-hidroxilasa y la alteración R384X a nivel del exón 7 en estado heterocigota, un codón stop prematuro que predice la presencia de una proteína truncada sin actividad biológica (Figs. 1 - 3). La madre presentó la mutación Q356X en estado heterocigoto.
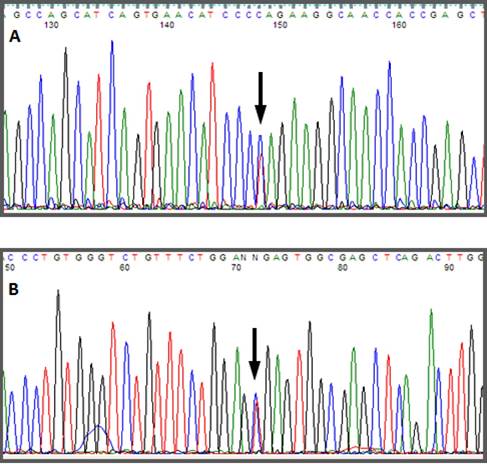
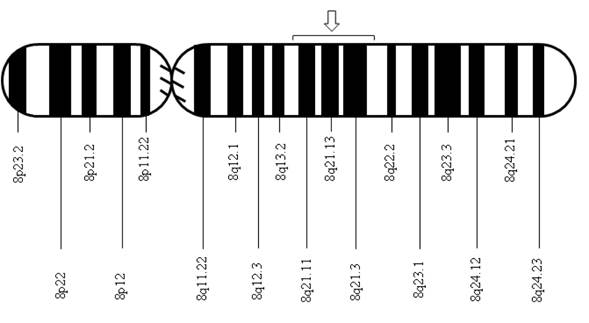
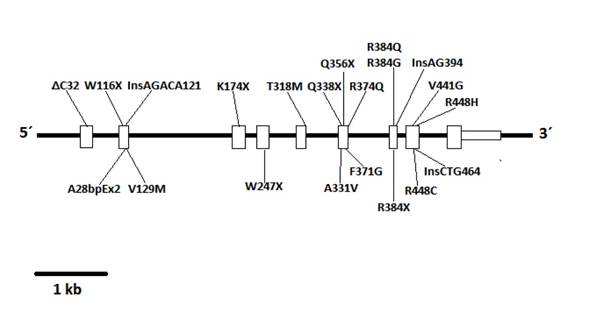
Intervención terapéutica
Una vez realizado el diagnostico se explicó a los padres sobre la complejidad y consecuencias que podría tener en el desarrollo del paciente esta patología, ellos deciden conservar su asignación de sexo como masculina, además se inicia manejo con prednisona a 14 mg/m2/día para buscar disminuir el estímulo de ACTH y frenar la producción de la 11-desoxicorticosterona (DOCA), buscando así el control de la tensión arterial (TA). Se debió utilizar propanolol para el control de la TA, inicialmente a 4 mg/Kg/día y finalmente a 40 mg/día debido a falta de adherencia y la no consecución de metas de TA. Tras la reducción de acné y vello púbico, junto con el descenso de la 17-OH progesterona, se decidió instaurar esquema de manejo con prednisona a 5 mg/día.
Seguimiento y resultados
El manejo farmacológico del paciente se vio limitado por la poca adherencia al tratamiento, lo que llevó al paciente a necesitar hospitalización un año después de su primera consulta, por presentarse en urgencia hipertensiva asociada a cefalea, posterior a esta hospitalización la adherencia mejoro y se logró optimizar el manejo, se retiró el propanolol y continuo con prednisona. En el seguimiento no se evidenció ningún trastorno electrolítico, el Tanner y la edad ósea se mantuvieron estables y los niveles de 17-OH progesterona descendieron de 52.0 a 28.0 ng/dL tras un año y tres meses de iniciado el tratamiento. Fue valorado por última vez en dos años después de la primera visita y se evidenció un descenso del acné, sin embargo persistía con pene adulto y Tanner 3.
Discusión
Hiperplasia adrenal congénita (HAC) se denominan a un grupo de patologías caracterizadas por el defecto de diferentes enzimas participes en la síntesis de esteroides adrenales, secundaría a la mutación de los diferentes genes que las codifican, que lleva a una hipersecreción de hormona liberadora de corticotropina (CRH) y ACTH 9. Su prevalencia a nivel mundial es de 1 por cada 100,000 personas 3, sin una predilección por algún sexo 1.
La HAC engloba un variado grupo de patologías, cada una determinada por una afección enzimática distinta y por grados diferentes de penetrancia. Entre las enzimas afectadas se encuentran la 11-beta-hidroxilasa, encargada del paso de desoxicorticosterona a corticosterona, ambas hormonas de acción mineralocorticoide y precursoras de la aldosterona, y de deoxicortisol a cortisol 10.
Su cuadro clásico se define por la ausencia de cortisol que lleva a un exceso en el estímulo de ACTH con acumulo de andrógenos y desoxicorticosterona (DOCA), por lo que los pacientes se presentan con alteraciones en el desarrollo sexual, como genitales ambiguos en el caso de las mujeres, pene grande para la edad en hombres, pubertad precoz o talla alta, además, el acumulo de mineralocorticoides puede llevar al desarrollo de trastornos hipertensivos, así como desbalances hidroelectrolíticos de patofisiología poco clara en las formas más severas 1,2,11. En el caso presentado, se encontró virilización casi total, talla alta y trastorno hipertensivo, sin alteración del estado hidroelectrolítico.
Acorde a su fisiopatología, se encontró en la bioquímica sanguínea, niveles de DOCA y deoxicortisol elevados, con disminución correspondiente de cortisol y aldosterona circulantes, y un nivel elevado de ACTH como consecuencia de la disminución de la acción del cortisol. Asociado a esto se pueden encontrar los niveles de andrógenos adrenales y testosterona moderadamente elevados y en ocasiones una elevación moderada de 17 OH-progesterona. También se ha descrito la supresión de secreción de renina secundario a la retroalimentación negativa por la acción mineralocorticoide aumentada, por la elevación de la DOCA 1,2. Llama la atención en el paciente, la presencia de niveles normales de cortisol y aldosterona, y niveles elevados de 17-OH progesterona, además con supresión de los niveles de renina y aumento de la androstenediona y de la DOCA, la cual sería finalmente la que mejor orientaría su diagnóstico. No se midieron niveles de deoxicortisol ni ACTH por inconvenientes en su disponibilidad en el medio local.
El diagnóstico genético se hizo a partir de la descripción de una mutación en el gen CYP11B1 10, como las múltiples previamente descritas 3,4,6-8. Una vez identificado el caso, se hace importante la caracterización genética de ambos padres de ser posible. En el caso reportado se encontró la mutación Q356X en estado heterocigoto tanto en madre como hijo, la cual había sido descrita en estado homocigoto, en pacientes de origen africano particularmente de Túnez 3,7, y una mutación R384X a nivel del exón 7 en estado heterocigoto en el paciente, la cual no había sido descrita previamente en la literatura. El gen R384X, al comportarse como un codón stop prematuro, predice la presencia de una proteína truncada sin actividad biológica la cual tendría una carga importante y sería la responsable de permitir el desarrollo de la patología a pesar de encontrarse la mutación Q356X en estado heterocigoto.
El tratamiento de la HAC está basado en la terapia de reemplazo hormonal a partir de la suplementación de glucocorticoides buscando como principal efecto cortar la síntesis de ACTH y así mismo su estímulo a nivel adrenal 12, en ocasiones se requiere manejo antihipertensivo específico muchas veces junto a diuréticos ahorradores de potasio 1. En el caso discutido, se inició manejo con Prednisona, con adecuada respuesta evaluada en niveles de 17 OH-progesterona, sin embargo la ausencia de un control adecuada de TA obligó al inicio de manejo de propanolol del cual necesitó múltiples ajustes de dosis llegando a requerir 40 mg/día. Muchas de las limitaciones en el control de síntomas a pesar del tratamiento fueron dadas por una pobre adhesión de la madre del paciente al manejo, motivo por el cual se podría atribuir la ausencia de control de TA con la monoterapia con glucocorticoide, por lo que se retiró el manejo antihipertensivo una vez se aseguró el adecuado cumplimiento al esquema esteroide por parte de la madre.
Conclusión
Se describe el caso de un paciente con una clínica secundaría al acumulo de DOCA, de diagnóstico tardío y no asociada a desequilibrio hidroelectrolíticos, quien hasta el momento tenía sexo asignado masculino a partir de la apariencia de sus genitales externos, quien apenas hasta los dos años de edad sería definido como XX. Adicionalmente, en el examen genético se encontró una mutación no descrita previamente en la literatura, la R384X, lo cual resalta la importancia de este caso. Cabe destacar la ausencia de testículos en el aparente escroto y canal inguinal al examen físico, lo cual de haber sido notificado desde el momento del nacimiento podría haber orientado a un diagnostico precoz y habría evitado el dilema de afrontar una discordancia entre sexo asignado y cromosómico a una mayor edad, por lo que se enfatiza en la importancia del examen físico y estudio pertinente del paciente con aparente anarquía.
Finalmente, las diversas mutaciones asociadas al déficit de 11-beta-hidroxilasa han sido asociadas a grupos étnicos y con características demográficas similares lo que arroja datos de las características de estas alteraciones, la descripción de una nueva mutación en Colombia, abre la posibilidad de descripciones del patrón genético de otros pacientes con deficiencia de 11-beta-hidroxilasa para determinar si es una mutación con efecto fundador, única en el país.
Agradecimientos:
Al Servicio de Endocrinología, Laboratorio de Biología Molecular Diagnóstico del Hospital de Pediatría Juan P. Garrahan y en especial a la Dra. Alicia Belgorosky, por su ayuda con el análisis genético del paciente.
REFERENCIAS
1. Nimkarn S, New MI. Steroid 11beta- hydroxylase deficiency congenital adrenal hyperplasia. Trends Endocrinol Metab. 2008; 19(3): 96-9. [ Links ]
2. Zachmann M, Tassinari D, Prader A. Clinical and biochemical variability of congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency. A study of 25 patients. J Clin Endocrinol Metab. 1983; 56(2): 222-9. [ Links ]
3. Curnow KM, Slutsker L, Vitek J, Cole T, Speiser PW, New MI, et al. Mutations in the CYP11B1 gene causing congenital adrenal hyperplasia and hypertension cluster in exons 6, 7, and 8. Proc Natl Acad Sci USA. 1993; 90(10): 4552-6. [ Links ]
4. Chua SC, Szabo P, Vitek A, Grzeschik KH, John M, White PC. Cloning of cDNA encoding steroid 11-beta-hydroxylase (P450c11). Proc Natl Acad Sci USA. 1987; 84(20): 7193-7. [ Links ]
5. Paperna T, Gershoni-Baruch R, Badarneh K, Kasinetz L, Hochberg Z. Mutations in CYP11B1 and congenital adrenal hyperplasia in Moroccan Jews. J Clin Endocrinol Metab. 2005; 90(9): 5463-5. [ Links ]
6. Soardi FC, Penachioni JY, Justo GZ, Bachega TA, Inácio M, Mendonça BB, et al. Novel mutations in CYP11B1 gene leading to 11ß-hydroxylase deficiency in Brazilian patients. J Clin Endocrinol Metab. 2009; 94(9): 3481-5. [ Links ]
7. Kharrat M, Trabelsi S, Chaabouni M, Maazoul F, Kraoua L, Ben Jemaa L, et al. Only two mutations detected in 15 Tunisian patients with 11ß-hydroxylase deficiency: the p.Q356X and the novel p.G379V. Clin Genet. 2010; 78(4): 1399-401. [ Links ]
8. Dumic K, Yuen T, Grubic Z, Kusec V, Barisic I, New MI. Two novel CYP11B1 gene mutations in patients from two croatian families with 11ß-hydroxylase deficiency. Int J Endocrinol. 2014; 2014(2014): 185974. [ Links ]
9. Antal Z, Zhou P. Congenital Adrenal Hyperplasia: Diagnosis, evaluation, and management. Pediatr Rev. 2009; 30(7): e49-57. [ Links ]
10. Peter M. Congenital adrenal hyperplasia: 11beta-hydroxylase deficiency. Semin Reprod Med. 2002; 20(3): 249-54. [ Links ]
11. Reisch N, Högler W, Parajes S, Rose IT, Dhir V, Götzinger J, et al. A diagnosis not to be missed: Non-classic steroid 11ß-hydroxylase deficiency presenting with premature adrenarche and hirsutism. J Clin Endocrinol Metab. 2013; 98(10): 1620-5. [ Links ]
12. German A, Suraiya S, Tenenbaum-Rakover Y, Koren I, Pillar G, Hochberg Z. Control of childhood congenital adrenal hyperplasia and sleep activity and quality with morning or evening glucocorticoid therapy. J Clin Endocrinol Metab. 2008; 93(12): 470710. [ Links ]
Recibido: 16 de Octubre de 2014; Revisado: 28 de Abril de 2016; Aprobado: 19 de Agosto de 2016
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
