











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkIntroduction
Pelizaeus Merzbacher Disease (PMD) is a chronic pediatric leukoencephalopathy caused by disorders of the axonal myelination and the myelin metabolism in the oligodendrocytes, reported for the first time on 1885 by doctor Friedrich Pelizaeus 1 and revisited on 1910 by Ludwig Merzbacher 2. Its genetic etiology affects the expression of the Proteolipidic Protein type 13,4, varying from hemizygous mutations to gene dosage alterations of the PLP1 (Xq22). Given the location of the causal gene, PMD is inherited in a X-linked recessive manner3.
Although clinical manifestations are heterogeneous 5,6, the most relevant neurological signs are nystagmus, developmental delay, spasticity, along with neuroimaging supporting aberrant myelination of the Central Nervous System (CNS) compromising primarily the periventricular white matter, with a tigroid striation pattern that responds to the conservation of myelinated islets, and also an alteration of the N-acetyl aspartate and choline profiles on the brain magnetic resonance spectroscopy 5,7.
Unlike other leukodystrophies in which there is a period of normal cortical myelination an then comes a disruption resulting in the lost of myelin sheaths (demyelination), PMD has, from the beginning, an abnormal or low production of this very important protein (hypomyelination), due to a damage on the PLP1 gene coding for the Protelipidic Protein type 1 that interferes with the oligodendrocyte synthesis of fully functional myelin sheaths and probably also affects the peripheral function of myelinated axons 3,8.
PMD corresponds to a larger group of neurological phenotypes known as PLP1 related disorders, all being allelic diseases: Connatal PMD, Classic PMD, Nule Syndrome (NS), Complicated Hereditary Spastic Paraplegia type 2 (SPG2) and Uncomplicated Spastic Paraplegia type 2, ranging in a wide variety of clinical manifestations which variability is not yet completely understood 5,9.
In general, PLP1 gene duplications result in a classical form of PMD, nonsense mutations in either form of SPG2 and connatal form of PMD, and other monoallelic mutations have been related to less circumscribed clinical phenotypes 6. Patients suffering from a connatal form of PMD, the most severe phenotype, have histopathological studies revealing complete absence of myelination in the brain, explaining the rapid clinical deterioration and suggesting tan death of these patients may respond to nervous conduction alterations in brain control centers. There’s also a phenotype of patients with clinical and radiological traits almost identical to those in PMD, with no PLP1 mutations detected, classified as PMD like (PMDL) syndrome 9.
Frequently, the connatal form of PMD is expressed during the first weeks of life, through key findings in the clinical neurological examination, that include pendular nystagmus, hypotonia and laryngeal stridor; later in life, seizures and sever motor deficits appear, and hypotonia turns to weakening limb spasticity; affected patients may never walk 10. Verbal language is limited, but patients understand simple orders and can follow them. Affected individuals with the connatal form of PMD die in infancy, usually secondary to respiratory or deglutition complications, such as bronchoaspiration 9.
Classic PMD is characterized in the first stages of disease by nystagmus, hypotonia and tremor in male affected patients, joint progressively by ataxia and spastic quadriparesis in the school age. Motor impairment of the limbs is less severe tan that presented in the connatal form, and patients can frequently achieve walking even if requiring special aids, and have better control of voluntary movement of the upper limbs 9,11. Classic PMD affected males also have improved cognitive development, with acceptable speech. Survival rates in this patients have been described to be up to the seventh decade of life.
On the other hand, NS patients suffer from a less harmful condition also caused by large deletions or damaging mutations resulting in loss of PLP1 protein product. As the phenotype is thought to be less severe than the other forms of PMD, some case series have even considered NS to be another variant of Complicated SPG2 9. Interestingly, it has been described NS affected individuals to have a multifocal demyelinating neuropathy 8,12 sometimes being the only clinical feature of the syndrome; NS patients do not present with nystagmus, their spastic paraplegia is mild, affecting primarily the lower limbs, and ataxic compromise may vary. Another differential diagnosis to consider is SPG2, an allelic disorder to PMD and NS, consisting of an heterogeneous constellation of clinical phenotypes primarily characterized by weakening and progressive lower limb spasticity during the first decade of life, with previous normal motor development. Patients can also have nystagmus, optic atrophy, dysarthria, ataxic features and variable range of intellectual disability; however, symptoms appear to be less compromising tan those presenting in classic PMD. Most of mutations detected on individuals diagnosed with SPG2 are missense 8. Also, it is worth mentioning that SPG2 affected males can reproduce, while there are no reports of PMD affected males who have descendants 9.
This article describes seven Colombian individuals with clinical, paraclinical and molecular diagnosis of PMD, through phenotype and gene variant characterization.
Ethical approval
Written informed consent was obtained from patient’s parents / legal guardians for publication of this report. Copies of the written consents are available for review by the Editors of this journal and are kept within the clinical records of each patient.
This study was approved by the ethics committee of the Faculty of Medicine of Universidad Nacional
Case series presentation
Seven individuals ages 6 months to 16 years (4 probands, 3 male relatives of the probands), diagnosed clinically, paraclinically and molecularly as Pelizaeus Merzbacher patients, attended in different medical care centers in Colombia (Fig. 1). They underwent clinical evaluations, neuroimaging (i.e. brain MRI), electro diagnosis (evoked visual and auditory potentials, electromyography, neural conduction velocity tests, computerized testing of gait), biochemical testing (i.e. blood and urine amino acids) and genetic testing (karyotyping, deletion/duplication analysis of PLP1 or whole gene sequencing of PLP1, targeted mutation studies). Also, we applied the PMD functional disability scoring system and the WHO Abbreviated Scale of Development to assess the degree of developmental retardation and disability on our patients 13.
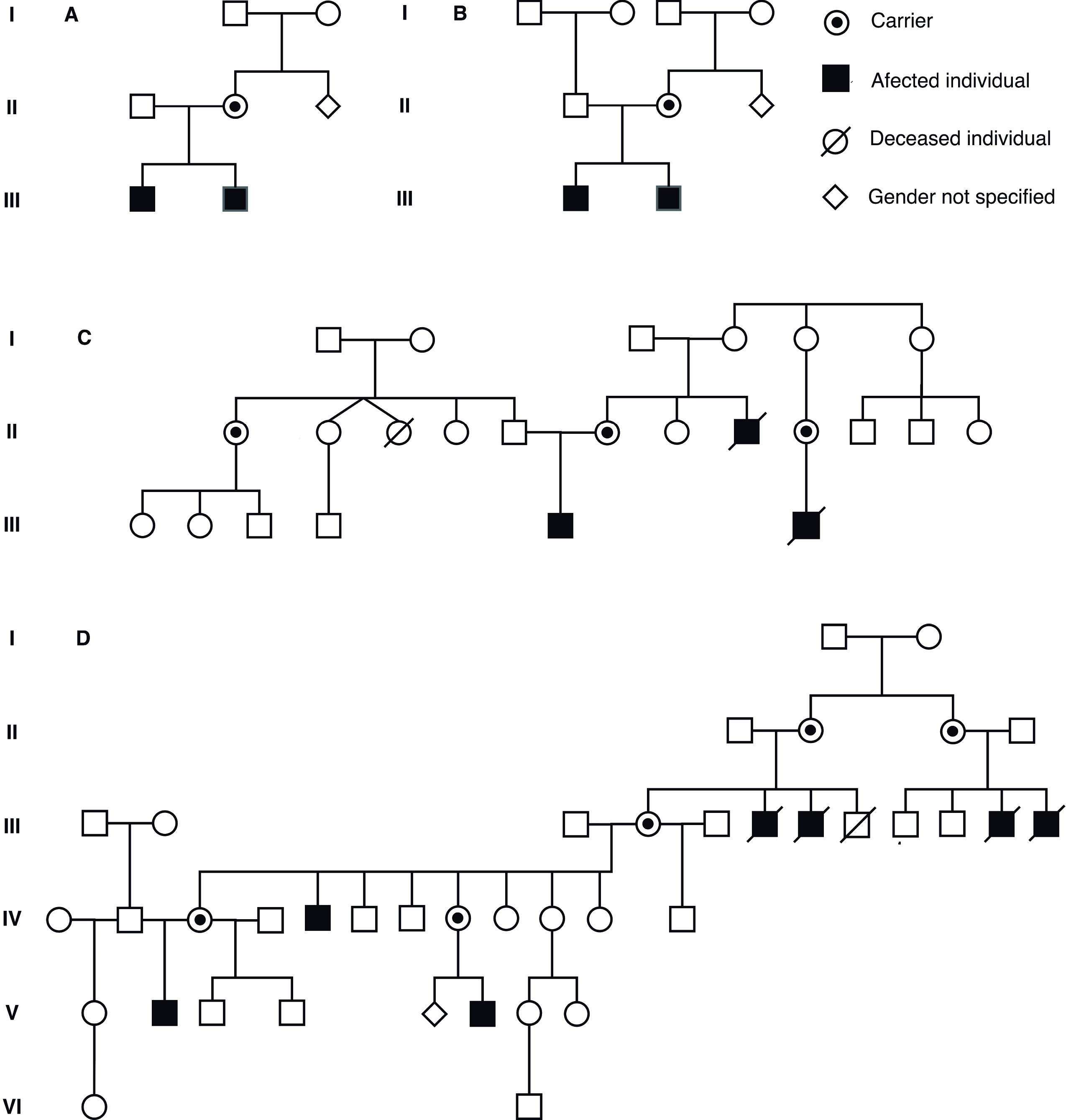
Figure 1 Pedigrees of families of the participant individuals. Note that patients are cited on the tables with their assigned pedigree numbers.
Methodologies used for the genetic testing of PLP1 gene were either multiplex ligation-dependent probe amplification (MLPA) using SALSA P022 A1 or B1 and P071 kits (MRC-Holland), which tested for deletions or duplications, whole gene sequencing by Sanger method for the detection of point mutations or small in/dels, or QT-PCR to confirm the presence of family mutations. In order to evaluate the impact of the molecular alterations detected, we used software as PolyPhen - 2 v.2.1 (http://genetics.bwh.harvard.edu/pph2/), HumVar model (http://genetics.bwh.harvard.edu/pph2) and SIFT (http://sift.jcvi.org) to predict pathogenic or benign changes on the PLP1 proteic product, having as reference sequence the one published under the entry NM_000533.3 (NCBI RefSeq, http://www.ncbi.nlm.nih.gov/nuccore) in the NCBI public databases.
Results
All patients were male, 6 months to 16 years of age, one of them died by the age of 5 due to complications of a respiratory infection. Mean age of onset of symptoms was 8 months, presenting with developmental delay on 100% of the cases and early onset nystagmus in 28.7%. Mean age at diagnosis was 5 years 5 months, being classic PMD most frequently diagnosed, in five cases, whereas the connatal phenotype was only present in two of the patients, (28.5%).
In our sample, two patients had history of cerebral palsy, being an actual comorbidity in only one of them. It is worth saying that all patients exhibited some level of speech delay or learning difficulties, and that only two were going to school.
In the physical examination, 57.0% of the patients had horizontal nystagmus while the others had the classic rotatory phenotype; none of them had oculomotor palsy or optic nerve atrophy; three individuals had any degree of sensorineural hearing loss. 57.0% showed signs of hypotonia, 28.5% an abnormal movement disorder, 71.4% any cerebellar signs, 85.7% any degree of spasticity, annotating that only three achieved gait with evident difficulties.
As for the two patients who had diagnosis of connatal PMD, it was documented both had experienced swallowing or deglutory disorders, history of seizures, microcephaly (in just one of them) and maturational ages in danger zones according to the WHO Abbreviated Scale of Psychosocial Development (WHO Abbreviated Scale of Psychosocial Development, https://www.unicef.org/bolivia/integrated_local_development_1480.htm).
When testing them for the PMD functional disability scoring system, all seven individuals had any level of disability, being moderate in 57.0% of the patients (10 to 20 points) or severe in 28.5% (under 10 points); it was not possible to evaluate the score on one patient given his very young age (Clinical endpoints shown in Table 1).
Table 1 Results from the clinical evaluation of patients with Pelizaeus Merzbacher Disease.
| Clinical features | 1. III - 1 | 1. III - 2 | 3. III - 7 | 2. III - 2 | 2. III - 1 | 4. V - 2 | 3. III - 6 |
|---|---|---|---|---|---|---|---|
| Age at onset | 2 yr. | 14 ms. | 6 ms. | 5 yr. | 1 yr. | 6 ms. | 0 ms. |
| Age at diagnosis | 5 yr 11 ms. | 2 yr y 8 ms. | 1.5 yr. | 9 yr. | 12 yr. | 7 yr. | 3 ms. |
| First symptoms | No head support or crawling. | DD. Nystagmus. | Hypotonia. DD. | DD. | DD. | No head support or crawling. | Laryngeal stridor. Nystagmus. |
| DD/ID | (+) | (+) | (+) | (+) | (+) | (+) | (+) |
| Nystagmus | Horizontal. | Horizontal. | Rotatory. | Rotatory. | Rotatory. | Horizontal. | Horizontal. |
| Language alteration | (+) | (+) | (+) | (+) | (+) | (+) | NA. |
| Spasticity | (+) | (+) | (+) | (+) | (+) | (+) | (-) |
| Walking | (-) | (-) | (-) | (+) | (+) | (+) | NA. |
| Swallowing issues | (-) | (-) | (+) | (-) | (-) | (-) | (+) |
| Hypotonia | (-) | (+) | (+) | (-) | (-) | (+) | (+) |
| Dysmetria/ Dysdiadochokinesia / Ataxia | (+) | (+) | (-) | (+) | (+) | (+) | (-) |
| PMD disability scoring system | 8 PTS. | 11 PTS. | 2 PTS. | 14 PTS. | 18 PTS. | 13 PTS. | NA. |
| WHO ASC | 72 PTS. | 94 PTS. | 29 PTS. | 115 PTS. | 86 PTS. | 100 PTS. | 25 PTS. |
Yr: Year. Ms: Months. DD: Developmental delay. ID: Intellectual disability. NA: Not applicable. PTS: Points. WHO ASC: World Healt Organization Abbreviated Scale of Development. (+): Present. (-): Absent.
Neuroimaging of patients with classic PMD showed evidence of T2 hyperintensities both diffuse or periventricular in the supratentorial withe matter. Other encephalic structures such as the brainstem, basal nuclei and cerebellum showed no abnormalities. In the connatal form affected individuals, we also observed hypo intensities of the basal nuclei and grey matter atrophy. 42.8% of the patients presented abnormal evoked auditory potentials and 28.5% abnormal evoked visual potentials; only one patient had abnormal neuro conduction velocities en another one had high levels of Mio inositol when tested for brain spectroscopic patterns.
Molecular studies were used in the majority of the cases to confirm the diagnosis. For two cases molecular confirmation was not considered necessary given their affected male brothers had already been tested. PLP1 gene dosage alterations (duplications) were found in 28.5% of the patients (two siblings), whereas three different single nucleotide variants were detected: c.140T>C (p.I47T), a missense variant classified as pathogenic, and two previously unreported alterations, the c.609C>T (p.Q99X) nonsense variant in two patients, and in one patient the c.152T>A (p.F51Y) missense variants. Laboratory endpoints are shown in Table 2.
Table 2 Results from the paraclinical evaluation of patients with Pelizaeus Merzbacher Disease.
| Paraclinical evaluation | 1. III - 1 | 1. III - 2 | 3. III - 7 | 2. III - 2 | 2. III - 1 | 4. V - 2 | 3. III - 6 |
|---|---|---|---|---|---|---|---|
| Connatal absence or myelin on MRI | (-) | (-) | (+) | (-) | (-) | (-) | (+) |
| MRI Hyperintensities localization | SC. PV. | Diffuse. | Diffuse. | PV. | PV / PaV. | PV. / PaV. | Diffuse. |
| Cerebral Spectroscopy | RP. | Normal patterns. | PV white matter irregular signal on T2. | Normal patterns. | Normal patterns. | PV posterosuperior white matter irregular signal on T2. | Normal patterns. |
| Auditory Evoked Potentials. | Auditory desynchrony. Type A tympanometry. | Auditory desynchrony vs. Auditory neuropathy. Severe compromise of physiological thresholds. | Normal patterns. | Normal patterns. | Normal patterns. | Normal patterns. | Normal patterns. |
| Visual Evoked Potentials. | Retrocorneal functional disorder. | Diffuse compromise of retinocorial pathways with axonal lost pattern. | Normal patterns. | Normal patterns. | Normal patterns. | Normal patterns. | RP. |
| NCV | Normal patterns. | Peroneal axonal neuropathy. | RP. | Normal patterns. | Normal patterns. | Normal patterns. | RP. |
| EMG | Normal patterns. | Normal patterns. | RP. | Normal patterns. | Normal patterns. | Normal patterns. | RP. |
| As worth spasticity index | UL 2 / IL 4. | UL 2 / IL 3. | UL / IL 4. | UL 1+ / IL 2. | UL 1+ / IL 1+. | RB 1+ / LB 1. | NA. |
| PLP1 molecular analysis | Complete duplication. | Complete duplication. | c.140T>C (p.I47T) hemizygous. | c.609C>T (p.Q99X) hemizygous. | c.609C>T (p.Q99X) hemizygous. | c.152T>A (p.F51Y) hemizygous. | c.140T>C (p.I47T) hemizygous. |
(+): Present. (-): Absent. MRI: Cerebral magnetic resonance imaging. NA: Not applicable. SC: Subcortical. PV: Periventricular. PaV: Paraventricular. RP: Results pending. NCV: Nerve conduction velocities. EMG: Electromyography. UL: Upper Limbs. IL: Inferior limbs.
Discussion
We present one of the first Latin-American series of patients with clinical diagnosis and molecular confirmation of Pelizaeus Merzbacher disease, being the classical form more frequent than the connatal form in the evaluated patients.
Developmental delay associated with nystagmus was key to diagnosis, both present in 100% of the cases. Along with the high clinical suspicion, supporting neuroimaging and molecular analysis permit an appropriate genetic counselling.
Connatal form of PMD is less frequent and far more severe than the classic phenotype. In our study, it is to note patients with the connatal form showed worse scores of disability (High severity scores in the PMD Disability Scoring System) and more pronounced developmental delay, and those continue to worsen until their deaths.
Diagnosis can be mistaken primarily with SPG2, also caused by mutations on the PLP1 gene, differing on signs such as autonomic dysfunction and characteristic paraplegia. NS, a variant of the PMD spectrum, presents as a periphery demyelinating neuropathy. Among other differential diagnosis we can count Krabbe disease, Canavan disease, other leukodystrophies and cerebral palsy. Connatal form of PMD is more severe than the classical form, a verifiable fact in our series, and with a reported expectancy of life lower than the first decade of life.
Point mutations were more frequently found, disregarding previous reports where >50% of PLP1 alterations are duplications. Case series have reported point mutations as the etiology of PMD related phenotypes in nearly 30% of male affected patients, yet we report them to be present in 71.5% of our cases and cannot rule out a signature genetic background for Latin-American patients with PMD, despite simple size (Scheme of mutations found in Figure 2).
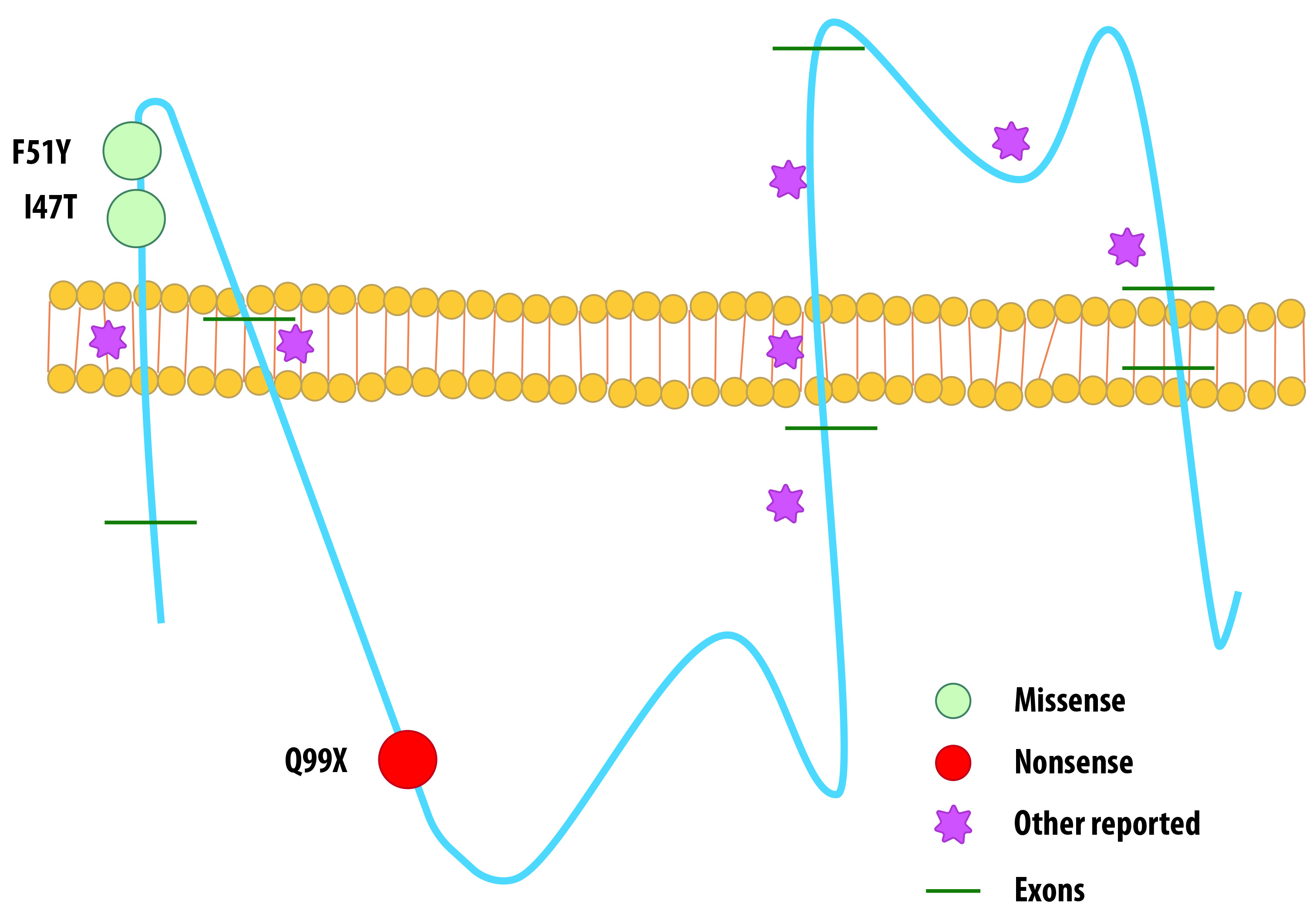
Figure 2 Schematic view of mutations found in our patients and previously reported mutations affecting PLP1 protein.
PLP1 gene is located in chromosomal region Xq22, with a 17 kb length, 7 exons and 6 introns. Exon 1 only transcribes the start codon, while exons 2, 3, 4, and 5 encode the hydrophobic domains and the hydrophilic chains of the protein. C-terminal transmembrane domain is encoded by exons 6 and 7. Besides the C-terminal hydrophilic domain, exón 7 contains the 3’UTR 14,15. PLP1 is further translated into the proteolipid protein 1 (PLP1), a 276 aminoacid peptide, or the isoform called DM20, which loses 35 residues inside its intracellular loop. Both PLP1 and DM20 are highly hydrophobic membrane proteins, accounting for up to 50% of compact myelin proteins in the central nervous system of the adult 16. PLP1/DM20 exact way of functioning has not been described precisely to date; however, it is clear they are needed for assembly and stability of the myelin sheath, and as before mentioned, PLP1 mutations have been widely studied as cause of PMD and SPG2. Studying male affected patients and animal models has led us to defy PLP1/DM20 actively participate in the synthesis of myelin intraperiod line, myelin compaction, myelin sheath adhesion to oligodendrocyte membrane, etc. A wide range of mutations in PLP1 has been described, recurrently detecting a whole gene duplication as the most frequent alteration 17,18,19.
Mutation c.140T>C found in our patients with the connatal form of the disease has already been reported by Hoffmann et al. in patients with classic PMD 5. Grossi et al.20, reported a similar mutation in a patient with a classic phenotype, an exon 2 microduplication (c.134_140dup7) that caused a frameshift (p.Ile47IlefsX4) and resulted in a truncated protein product, 4 amino acids downstream 21. We believe it is important to establish the biochemical functionality of I47 position on the myelin proteolipidic protein to evaluate its impact on the connatal phenotype of PMD disease, given that there are not functional studies to this date that prove in vitro or in vivo effects.
Nonsense mutation c.609C>T (p.Q99X) and missense mutation c.152T>A (p.F51Y) have not been previously reported as causes of PMD disease, but both their SIFT and PolyPhen scores suggest they are damaging (0.82 and 0.98 Polyphen scores respectively). This variants express as a compromise of two functional domains of the PLP1 protein: c.609C>T (p.Q99X) affects the cytoplasmic domain while c.152T>A (p.F51Y) affects an extracellular topological domain 14,16.
In a smaller percentage of cases triplications and other dosage alterations in the PLP1 gene have been reported, and less than 2% of cases so far reported have shown a complete or partial deletion of the gene 22-24.
Conclusions
To our knowledge, this is not only one of first Latin-American case series but the larger one, presenting the main characteristics of the clinical diagnosis and molecular signatures of PMD male affected patients, being the classical form overall more frequent than the connatal form. Both patients with the connatal form of the disease had severe disability scores and poor vital prognosis, despite having the chance of an earlier diagnosis.
According to our results, we propose that for our population the diagnostic algorithm begins on a high clinical suspicion, followed by paraclinical extension in which neuroimaging is crucial, moving on to the molecular confirmation by using approaches to simultaneously sequence the PLP1 gene in order to detect point mutations and in/dels and performing a deletion/duplication analysis for the detection of gene dosage alterations.
In spite of the incapacitating character of this disease, patients with less severe or moderate forms of PMD have rather normal life expectancy, but there are records of patients with severe classical forms who died past the second decade of life. Because of this, it is a priority for the clinical specialists and treating physicians to improve the diagnosis algorithms in order to shorten time before establishment of the specific therapeutic plan and the appropriate genetic counselling for the families.

