










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkColombia Médica
On-line version ISSN 1657-9534
Colomb. Med. vol.49 no.2 Cali Apr./June 2018
https://doi.org/10.25100/cm.v49i2.2522
Reporte de caso
Heterogeneidad clínica y mutacional en pacientes colombianos con Pelizaeus Merzbacher
1 Maestria en Genética Humana, Facultad de Medicina, Universidad Nacional de Colombia, Bogotá, Colombia.
2 Hospital Militar Central, Bogotá, Colombia.
3 Instituto de Ortopedia Infantil Roosevelt, Bogotá, Colombia.
4 Genetica Medica, Facultad de Medicina, Pontificia Universidad Javeriana. Bogotá, Colombia
5 Hospital La Victoria, Bogotá, Colombia.
Descripción del caso:
La enfermedad de Pelizaeus Merzbacher es una leucodistrofia ligada al X que causa encefalopatía espástica crónica en la infancia. Su etiología es genética, por duplicaciones u otros trastornos de la dosis génica o mutaciones puntuales del gen PLP1, lo que condiciona la formación anormal de las vainas de mielina principalmente en el sistema nervioso central. Clínicamente se caracteriza por un cuadro de retardo del neurodesarrollo, nistagmus y espasticidad, con neuroimágenes que evidencian la dismielinización. Presentamos una serie de siete casos colombianos con esta leucodistrofica en la que describimos fenotípica y genotípicamente la heterogeneidad de esta enfermedad en nuestra población.
Hallazgos clínicos:
Todos los pacientes analizados fueron de sexo masculino, con edad promedio de inicio de síntomas hacia los ocho meses de vida. La edad media al diagnóstico fue de 5 años 5 meses, siendo más frecuente el diagnóstico de PMD clásica que el tipo connatal. Se describe retardo del desarrollo motor en el 100% de los casos, acompañado de nistagmus en el 28.7%. 85.7% de los casos tenía algún grado de espasticidad, 71.4 % signos cerebelosos, 57.0% hipotonía, y hasta en 28.5% se evidenciaron movimientos anormales. Solo tres pacientes lograron marcha, aunque patológica. En los dos pacientes con la forma connatal se documentó una edad maduracional motora en el rango de Alerta, de acuerdo a la escala abreviada del desarrollo de la OMS. En todos los casos se detectó algún tipo de anormalidad en el estudio imagenológico cerebral.
Estudios Moleculares y Resultados:
El diagnóstico molecular se empleó en la mayoría de los casos (85.7%), encontrando alteraciones en la dosis génica en el 28.5% y tres diferentes mutaciones puntuales.
Relevancia clínica:
Dados los hallazgos en los resultados del estudio molecular, sugerimos que en el abordaje diagnóstico confirmatorio para la población colombiana se debería contemplar en un mismo tiempo tanto la secuenciación como el estudio de variantes del número de copias del gen afectado, contrario a lo sugerido en literatura mundial en la que se inicia con estudio para duplicación / deleción.
Palabras clave: Enfermedad de Pelizaeus Merzbacher; desarrollo infantil; discapacidades del desarrollo; vaina de mielina; proteína proteolipídica de la mielina
Case Presentation:
Pelizaeus Merzbacher Disease (PMD) is an X-linked developmental defect of myelination that causes childhood chronic spastic encephalopathy. Its genetic etiology can be either a duplication (or other gene dosage alterations) or a punctual mutation at the PLP1 locus. Clinically, it presents with developmental delay, nystagmus and, spasticity, supported by neuroimaging in which the defect of myelination is evident. We present a series of seven Colombian patients diagnosed with this leucodystrophy, describing their genotypic and phenotypic characteristics and heterogeneity.
Clinical Findings:
All patients included were male, 6 months to 16 years of age. Mean age at onset of symptoms was 8 months. Mean age at diagnosis was 5 years 5 months, being classic PMD most frequently diagnosed, as compared to the connatal phenotype. All cases had a primary diagnosis of developmental delay on 100%, and in 28.7% of cases, early onset nystagmus was described. 85.7% of patients had spasticity, 71.4% cerebellar signs, 57.0% hypotonia, and 28.5% had an abnormal movement disorder. Only three patients were able to achieve gait, though altered. In the two patients who had a diagnosis of connatal PMD maturational ages in danger zones according to the WHO Abbreviated Scale of Psychosocial Development were documented. All cases had abnormalities in neuroimages.
Molecular Analysis and Results:
Molecular studies were used in the majority of the cases to confirm the diagnosis (85.7 %). For two cases molecular confirmation was not considered necessary given their affected male brothers had already been tested. PLP1 gene dosage alterations (duplications) were found in 28.5 % of the patients (two siblings), whereas three different single nucleotide variants were detected.
Clinical Relevance:
According to these findings, as authors we propose the diagnostic algorithm in Colombian population to begin on a high clinical suspicion, followed by paraclinical extension, moving on to the molecular confirmation by using approaches to simultaneously sequence the PLP1 gene in order to detect point mutations and in/dels and performing a copy number variation analysis for the detection of gene dosage alterations.
Keywords: Pelizaeus Merzbacher Disease; Developmental Disabilities; Child Development; Myelin Sheatn; Myelin Proteolipidic Protein
Introducción
La leucodistrofia o enfermedad de Pelizaeus Merzbacher (PMD) es una encefalopatía crónica de la infancia causada por desórdenes de la mielinización axonal y del metabolismo de la mielina oligodendroglial, descrita por primera vez en 1885 por Pelizaeus 1 y estudiada a mayor profundidad en 1910 por Merzbacher 2. Su etiología es genética por mutación hemicigota o alteraciones en la dosis génica del gen PLP1 (Xq22), que afectan la expresión de la Proteína Proteolipídica de la Mielina 1 3,4. Dada la localización y función del gen causal, PMD presenta una herencia ligada al cromosoma X recesiva 3.
Aunque las manifestaciones clínicas son muy heterogéneas 5,6, los signos clínicos primarios neurológicos incluyen nistagmo, retardo global del desarrollo, espasticidad, acompañados de características radiológicas vistas por Resonancia Nuclear Magnética cerebral (RM) como una mielinización del sistema nervioso central (SNC) aberrante de predominio periventricular sin afectación subcortical, junto a la presencia de islotes mielinizados que dan una apariencia ‘tigroide’ a la sustancia blanca, así como alteración de los patrones de N-acetil aspartato y colina en la espectroscopía que reflejan anomalías mielínicas axonales 5,7.
A diferencia de otras leucodistrofias en las que la mielina se produce pero es destruida posteriormente (desmielinizantes), en los pacientes con PMD la mielina no se produce de forma adecuada. Se cree que la hipo o dismielinización responde principalmente a que la afectación de la proteína proteolipídica mielínica afectada interfiere con la formación de las vainas de mielina de los oligodendrocitos en el SNC y con la formación de éstas vainas en los axones periféricos 3,8.
La PMD forma parte de un grupo más amplio de fenotipos neurológicos conocidos como trastornos relacionados con el gen PLP1, que son todos desórdenes alélicos: la forma connatal de PMD, la forma clásica de PMD, el síndrome nulo (SN), la Paraplejía Espástica tipo 2 (SPG2) complicada y la Paraplejía Espástica tipo 2 no complicada, todos ellos caracterizados por un diverso espectro de manifestaciones clínicas, cuya explicación de variabilidad no se comprende completamente 5,9.
En general, se considera que las duplicaciones del gen PLP1 dan lugar a la forma clásica de PMD, las mutaciones nonsense a las formas de Paraplejía Espástica complicada o no complicada (SPG2) y a la forma connatal de PMD y otras mutaciones monoalélicas se han relacionado con diferentes formas clínicas menos delimitadas 6. En los pacientes con la forma connatal de PMD, que es la más severa, el estudio histopatológico revela una ausencia completa de la mielinización en todo el cerebro, lo que podría explicar el rápido deterioro clínico y sugiere que la muerte, que en ocasiones es inexplicable, sea causada por alteraciones de la conducción nerviosa en los centros de control vital encefálicos. Existe un grupo de pacientes con características clínicas y neurorradiológicas casi idénticas a la PMD, en los que no se detectan mutaciones de PLP1 y que se clasifican como pacientes con un síndrome PMD like (PMLD) 9.
Clínicamente, la forma connatal de PMD se hace manifiesta en las primeras semanas de vida, por hallazgos al examen neurológico del recién nacido que incluyen nistagmos pendular, hipotonía, y estridor laríngeo; en edades más avanzadas aparecen crisis epilépticas y déficit motor severo y la hipotonía tiende a transformarse en espasticidad de las extremidades que puede ser muy limitante, con pacientes afectados que pueden nunca lograr la marcha o tener buena funcionalidad de sus miembros superiores 10. El lenguaje verbal es limitado, pero los pacientes comprenden órdenes sencillas. Los individuos afectados con esta forma de PMD mueren en la infancia, generalmente por complicaciones relacionadas con problemas deglutorios, como lo es la broncoaspiración 9.
La forma clásica de PMD descrita por Pelizaeus y posteriormente por Merzbacher, se caracteriza inicialmente por la aparición de nistagmos, hipotonía y tremor en los varones afectados, que se acompañan progresivamente de ataxia y cuadriparesia espástica. Lo anterior ocurre en los primeros años de vida. El compromiso motor de las extremidades es menos severo que en la forma connatal, logrando la marcha pero usualmente con requerimiento de dispositivos de apoyo y en porcentaje importante de los casos con un adecuado control voluntario de los miembros superiores 9,11. Los afectados también tienen mejor desarrollo cognitivo que aquellos que padecen la forma connatal, con lenguaje verbal aceptable. La supervivencia de los pacientes con esta forma de la enfermedad se ha descrito incluso hasta la séptima década de la vida.
Existe también una variante de PMD conocida como Síndrome Nulo (SN). Se trata de una condición causada por la presencia de grandes deleciones genómicas o mutaciones que resultan en la pérdida de las proteínas PLP1/DM20 en la que los pacientes presentan un único fenotipo clínico descrito como más leve que el causado por otras mutaciones en el mismo gen, y que en algunas series de pacientes es considerada como una forma complicada de SPG2 9. De manera interesante, se describe que estos pacientes presentan una neuropatía periférica desmielinizante multifocal 8,12, que incluso puede ser el único hallazgo del SN; los pacientes con SN no presentan nistagmos y la paraplejia espástica que padecen es usualmente leve, se asocia a variables grados de ataxia, y afecta principalmente a los miembros inferiores.
El segundo diagnóstico diferencial a tener en cuenta es la paraplejia espástica hereditaria de tipo 2 (SPG2), enfermedad alélica a PMD y el SN. SPG2 representa un grupo de desórdenes genéticamente heterogéneo con una amplia variabilidad clínica, en el que individuos que la padecen generalmente tienen desarrollo motor normal en los primeros años de vida, con inicio de debilidad más espasticidad progresiva de las extremidades inferiores entre los 2 y los 10 años de edad; los pacientes pueden presentar también nistagmos, atrofia óptica, disartria, ataxia, y grados variables de discapacidad intelectual en las formas complicadas; sin embargo, estos síntomas aparecen con menor severidad que en el fenotipo clásico de PMD. Las mutaciones hasta ahora descritas encontradas en individuos con diagnóstico de SPG2 son principalmente de sentido erróneo (missense) 8. Cabe anotar que se han reportado descendientes de varones con SPG2 pero no de varones con PMD 9.
En este artículo se describe a siete individuos con diagnóstico clínico, paraclínico y molecular compatible con PMD a partir de la descripción fenotípica y genotípica de esta enfermedad en pacientes colombianos.
Presentación de casos
La metodología utilizada para las pruebas genéticas del gene PLP1 fue la amplificación de sondas dependiente de ligandos múltiples (MLPZ) usando los kits SALSA P022 A1 o B1 y P071 (MRC-Holland), para probar delecion o duplicación y ña secuenciación del gen por el método de Sanger para la detección de mutaciones puntuales o in/del pequeños, o QT-PCR para confirmar la presencia de mutaciones familiares. Para evaluar el impacto de las alteraciones moleculares usamos los programas PolyPhen - 2 v.2.1 (http://genetics.bwh.harvard.edu/pph2/), HumVar model (http://genetics.bwh.harvard.edu/pph2) y SIFT (http://sift.jcvi.org) para predecir los cambios patogenicos o benignos en el produto proteico de PLP1, teniendo como referencia la secuencia de la publicación NM_000533.3 (NCBI RefSeq, http://www.ncbi.nlm.nih.gov/nuccore) en la base de datos publica de NCBI.
Resultados
Se incluyeron 7 pacientes con edades de 6 meses a 16 años (4 probandos, dos hermanos y un primo, todos afectados) con diagnóstico clínico, paraclínico y molecular de PMD, atendidos en diferentes centros asistenciales de Colombia. Las genealogías de los casos se muestran en la Figura 1. Dependiendo del subtipo de PMD (clásico o connatal), de la historia natural de la enfermedad y de sus diagnósticos diferenciales iniciales, los pacientes tuvieron acceso a neuroimágenes (RM cerebral simple / contrastada / espectroscopía), electrodiagnóstico (potenciales evocados auditivos, potenciales evocados visuales, electromiografía, velocidades de neuroconducción o test computarizado de marcha), pruebas bioquímicas (estudios diagnósticos para descartar aminoacidopatías o acidemias orgánicas, por ejemplo) y genéticas (cariotipo, estudio molecular de gen PLP1 y estudio de portadores), para poder determinar el diagnóstico final. Además se tuvo en cuenta las variables clínicas y paraclínicas para poder determinar el grado de severidad de la enfermedad empleando el PMD functional disability scoring system, el cual evalúa el impacto de la discapacidad en esta patología 13.
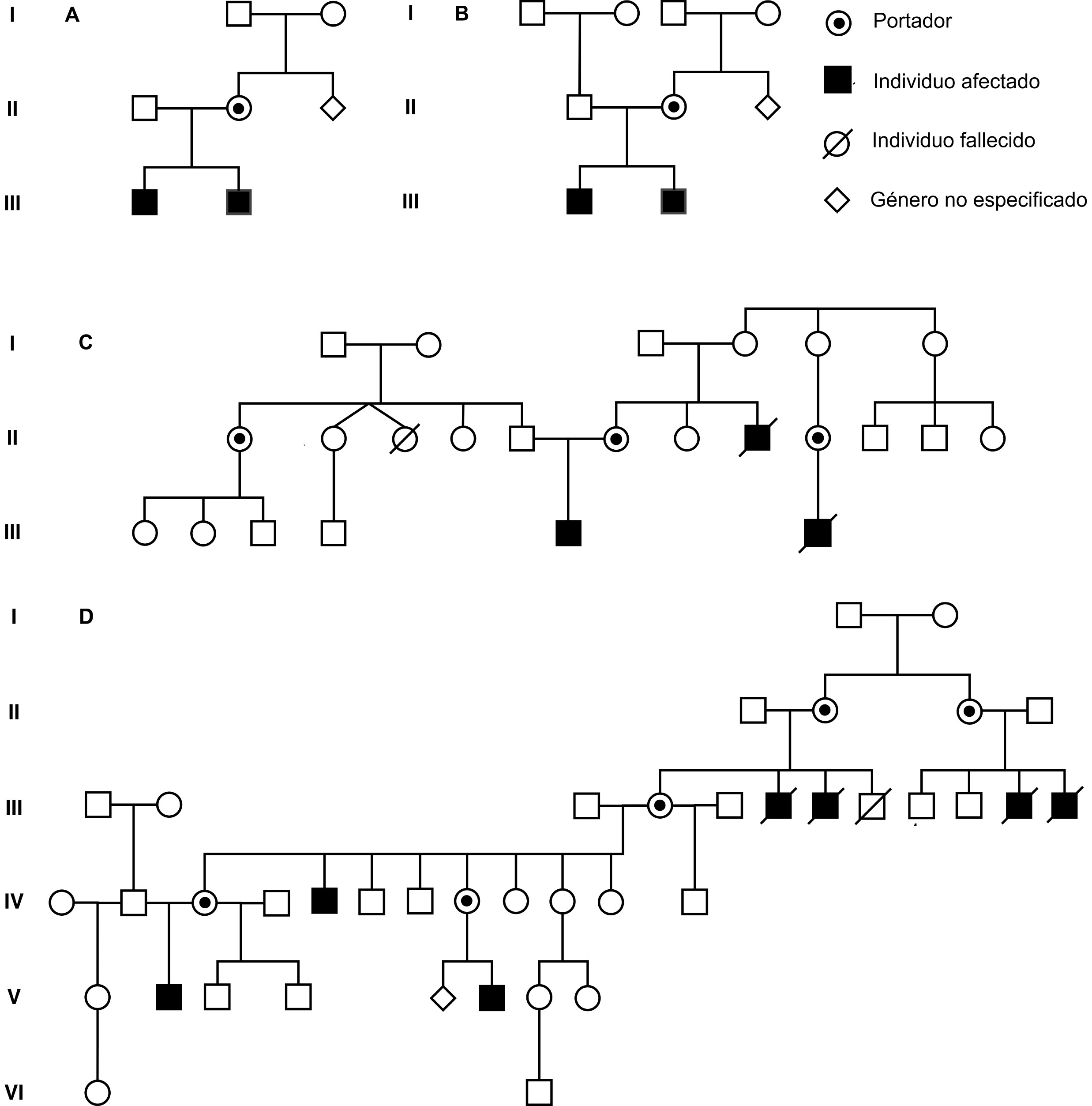
Figura 1 Genealogías de los casos presentados. Se incluyeron siete pacientes con diagnóstico clínico, paraclínico y molecular de PMD, en A. Los individuos III.1 y III.2, en B. Los individuos III.1 y III.2, en C los casos III.6 y III.7, en D el individuo V.2. Se demuestran las relaciones de consanguinidad entre algunos de ellos, comentadas en el texto: Dos parejas de hermanos, una pareja de primos, y un caso en quien no se conoció a otros familiares afectados.
Para los estudios moleculares se realizaron varios abordajes técnicos, tanto para el análisis de dosis génica como para la detección de mutaciones puntuales. En el primer caso se empleó el test de amplificación múltiple de sondas dependiente de ligamiento (MLPA por sus siglas en inglés), usando el SALSA P022 A1 o B1 y P071 (MRC-Holland), de acuerdo a las recomendaciones del proveedor o el protocolo de QT PCR cuantitava, y en un segundo abordaje se empleó secuenciación del gen PLP1, ya que el análisis por MLPA para deleción o duplicación fue negativo.
En el análisis de la variantes encontradas en el gen PLP1, se empleó la secuencia de referencia NM_000533.3 (NCBI RefSeq, https://www.ncbi.nlm.nih.gov/gene/5354) y para valorar el impacto de las mutaciones en la estructura proteica y la función se sometieron los resultados a diferentes programas de predicción bioinformática como PolyPhen - 2 v.2.1 (http://genetics.bwh.harvard.edu/pph2) y SIFT (http://sift.jcvi.org).
Resultados
Todos los pacientes analizados fueron de sexo masculino, con edades entre 6 meses a 16 años, uno de ellos fallecido a los 5 años de vida por infección respiratoria complicada. La edad promedio de inicio de síntomas fue de 8 meses, con retardo del desarrollo motor en el 100% de los casos, acompañado de nistagmos en el 28.7%. La media de edad de diagnóstico fue de 5 años 5 meses, siendo el diagnóstico de PMD clásica el más frecuente, con cinco pacientes, mientras el tipo connatal se encontró en dos individuos relacionados (28.5%).
En esta muestra, dos pacientes presentaron como antecedente, diagnostico de parálisis cerebral, estando realmente presente como comorbilidad en uno de ellos, lo cual se pudo confirmar por los hallazgos prenatales de su historia clínica. Adicional al retardo del desarrollo existente en esta muestra, todos los pacientes cursaron también con algún grado de retardo del lenguaje. De los 5 pacientes mayores de 5 años, solamente 2 se encontraban escolarizados y su rendimiento académico fue irregular.
Al examen físico de los pacientes descritos, el 57.0% cursaron con nistagmus horizontal y los 3 pacientes restantes con enfenotipo rotatorio clásico; ninguno presentó parálisis de oculomotores ni atrofia de nervio óptico y 3 pacientes tuvieron hipoacusia neurosensorial. El 57.0% presentaron hipotonía, el 28.5% movimientos anormales, el 71.4% signos cerebelosos, el 85.7% tenían algún grado de espasticidad. Cabe anotar que tres 3 pacientes lograron la marcha, aunque esta es fue patológica.
En los dos pacientes con la forma connatal se documentó dificultad para la alimentación, antecedentes de crisis convulsivas, en uno de ellos se encontró microcefalia y ambos tenían una edad maduracional motora en el rango de Alerta, de acuerdo a la escala abreviada del desarrollo de la OMS (WHO Abbreviated Scale of Psychosocial Development, https://www.unicef.org/bolivia/integrated_local_development_1480.htm).
Al aplicar el PMD functional disability scoring system sobre los 7 pacientes, todos presentaron puntajes de discapacidad moderada (10 a 20 puntos en el 57.0% de los pacientes) a severa (por debajo de 10 puntos en el 28.5%), excepto por un paciente en el que por su corta edad no fue posible evaluarlo. Los resultados del análisis clínico se encuentran resumidos en la Tabla 1.
Tabla 1 Resultados de la evaluación clínica de los pacientes con Pelizaeus Merzbacher.
| Caractersísticas clínicas | 1. III - 1 | 1. III - 2 | 3. III - 7 | 2. III - 2 | 2. III - 1 | 4. V - 2 | 3. III - 6 |
|---|---|---|---|---|---|---|---|
| Edad de inicio | 2 años | 14 meses | 6 meses | 5 años | 1 año | 6 meses | 0 meses |
| Edad al diagnóstico | 5 años 11 meses | 2 años y 8 meses | 1.5 años | 9 años | 12 años | 7 años | 3 meses |
| Primeros síntomas o signos | DD | DD. Nistagmos | Hipotonía. DD | DD | DD | DD | Estridor laríngeo. Nistagmos |
| DD/ID | + | + | + | + | + | + | + |
| Nistagmos | Horizontal | Horizontal | Rotatorio | Rotatorio | Rotatorio | Horizontal | Horizontal |
| Trastorno del lenguaje | + | + | + | + | + | + | NA |
| Espasticidad | + | + | + | + | + | + | - |
| Marcha | - | - | - | Autónoma, con dificultad | Autónoma, con dificultad | Autónoma, con dificultad | NA |
| Trastornos deglutorios | - | - | + | - | - | - | + |
| Hipotonía | - | + | + | - | - | + | + |
| Dismetría/ Disdiadoconesia / Ataxia | + | + | - | + | + | + | - |
| PMD disability scoring system | 8 PTS | 11 PTS | 2 PTS | 14 PTS | 18 PTS | 13 PTS | NA |
| WHO ASC | 72 PTS | 94 PTS | 29 PTS | 115 PTS | 86 PTS | 100 PTS | 25 PTS |
DD: Retardo del desarrollo. ID: Discapacidad Intelectual. NA: No aplica. PTS: Puntos. WHO ASC: World Healt Organization Abbreviated Scale of Development / Escala abreviada del desarrollo de la OMS. (+): Presente. (-): Ausente.
Las neuroimagenes de los pacientes con PMD clásico reportaron hiperintensidades tanto difusas como periventriculares de la sustancia blanca supratentorial. Las estructuras como el tallo cerebral, los núcleos basales y el cerebelo no presentaron alteraciones. En los casos de la forma connatal, se observó, además de lo descrito arriba, hipointensidades de los núcleos basales y atrofia de sustancia gris. El 42.8% presentaron alteraciones en los potenciales evocados auditivos y en el 28.5% en los visuales; un paciente mostró alteraciones en la neuroconducciones y en un caso, se observó un incremento de Mioinositol determinado en la espectroscopia por resonancia.
El diagnóstico molecular se empleó en la mayoría de los casos (85.7%), encontrando alteraciones en la dosis génica en el 28,5% y tres diferentes mutaciones puntuales, dos de ellas no reportadas previamente. Dos pacientes presentaron la duplicación completa del gen PLP1, otros dos presentaron la mutación misssense c.140T>C (p.I47T), en dos pacientes se encontró la mutación nonsense c.609C>T (p.Q99X) y otro paciente presentó la mutación missense c.152T>A (p.F51Y), estas dos últimas, privadas. Los resultados del análisis paraclínico de los casos se encuentran resumidos en la Tabla 2.
Tabla 2 Resultados de la evaluación paraclínica de los pacientes con Pelizaeus Merzbacher.
| Paraclínicos | 1. III - 1 | 1. III - 2 | 3. III - 7 | 2. III - 2 | 2. III - 1 | 4. V - 2 | 3. III - 6 |
|---|---|---|---|---|---|---|---|
| Ausencia connatal de mielina en la resonancia magnética | - | - | + | - | - | - | + |
| Localización de hiperintensidades en la resonancia magnética | SC. PV | Difusas | Difusas | PV | PV / PaV | PV /PaV | Difusas |
| Espectroscopia cerebral | SD | Patrones normales | Señal irregular de la materia blanca PV en T2 | Patrones normales | Patrones normales | Señal posterosuperior PV de materia blanca irregular en T2 | Patrones normales |
| Potenciales Evocados Auditivos | Desincronía auditiva. Timpanometría tipo A. | Desincronía auditiva vs. Neuropatía auditiva. Grave compromiso de los umbrales fisiológicos | Patrones normales | Patrones normales | Patrones normales | Patrones normales | Patrones normales |
| Potenciales Evocados Visuales | Trastorno funcional retrocorneal | Compromiso difuso de las vías retinianas con patrón e pérdida axonal | Patrones normales | Patrones normales | Patrones normales | Patrones normales | SD |
| VNC | Patrones normales | Neuropatía axonal peronea. | SD | Patrones normales | Patrones normales | Patrones normales | SD |
| EMG | Patrones normales | Patrones normales | SD | Patrones normales | Patrones normales | Patrones normales | SD |
| Índice de espasticidad de Asworth | MS 2 / MI 4 | MS 2 / MI 3 | MS / MI 4 | MS 1+ / MI 2. | MS 1+ / MI 1+. | MS 1+ / MI 1 | NA. |
| Estudio molecular de PLP1 | Duplicación completa del gen | Duplicación completa del gen | c.140T>C (p.I47T) hemicigoto | c.609C>T (p.Q99X) hemicigoto | c.609C>T (p.Q99X) hemicigoto | c.152T>A (p.F51Y) hemicigoto | c.140T>C (p.I47T) hemicigoto |
(+): Presente. (-): Ausente. RM: Resonancia Nuclear Magnética cerebral. NA: No aplica. SC: Subcortical. PV: Periventricular. PaV: Paraventricular. SD: Sin Dato. VNC: Velocidades de Neuroconducción. EMG: Electrommiografía. MS: Miembros superiores. MI: Miembros inferiores.
Discusión
Nosotros presentamos la serie mas grande de pacientes Latinoamericanos con diagnóstico clínico y confirmación molecular del espectro de Enfermedad de Pelizaeus Merzbacher, donde la presentación más frecuente se observó en la forma clásica, sin embargo también se lograron identificar dos pacientes con forma connatal.
Fueron claves para el diagnóstico en estos pacientes la presencia de un retardo global del desarrollo asociado a cualquier tipo de nistagmos, ambos signos estuvieron presentes en todos los pacientes. Fue necesario el diagnóstico molecular confirmatorio para el adecuado asesoramiento genético de las familias.
La forma connatal de la PMD es menos frecuente y mucho más severa que la forma clásica. En nuestro estudio fue notable que los pacientes con la forma connatal tienen mayor discapacidad (Puntajes en rangos de alta severidad en el PMD Disability Scoring System) y un más pronunciado retardo en el neuro desarrollo hasta el momento en que fallecen. Cabe mencionar que si bien no alcanzan rangos de alerta en la Escala Abreviada del Desarrollo, es probable que se deba a la temprana edad en que mueren.
En este estudio, en pacientes con diagnóstico de PMD fueron más frecuentes las mutaciones puntuales que las duplicaciones, contrario a lo reportado en literatura, donde el 50% de las alteraciones del gen PLP1 son defectos en la dosis génica (Fig. 2). Las series de casos de la literatura reportaron la aparición de mutaciones puntuales causales en un 30% de los varones con PMD, nosotros encontramos mutaciones puntuales en el 71.5%, probablemente debido al tamaño de la muestra, aunque no se debe descartar un background genético particular en latinoamericanos para esta patología.
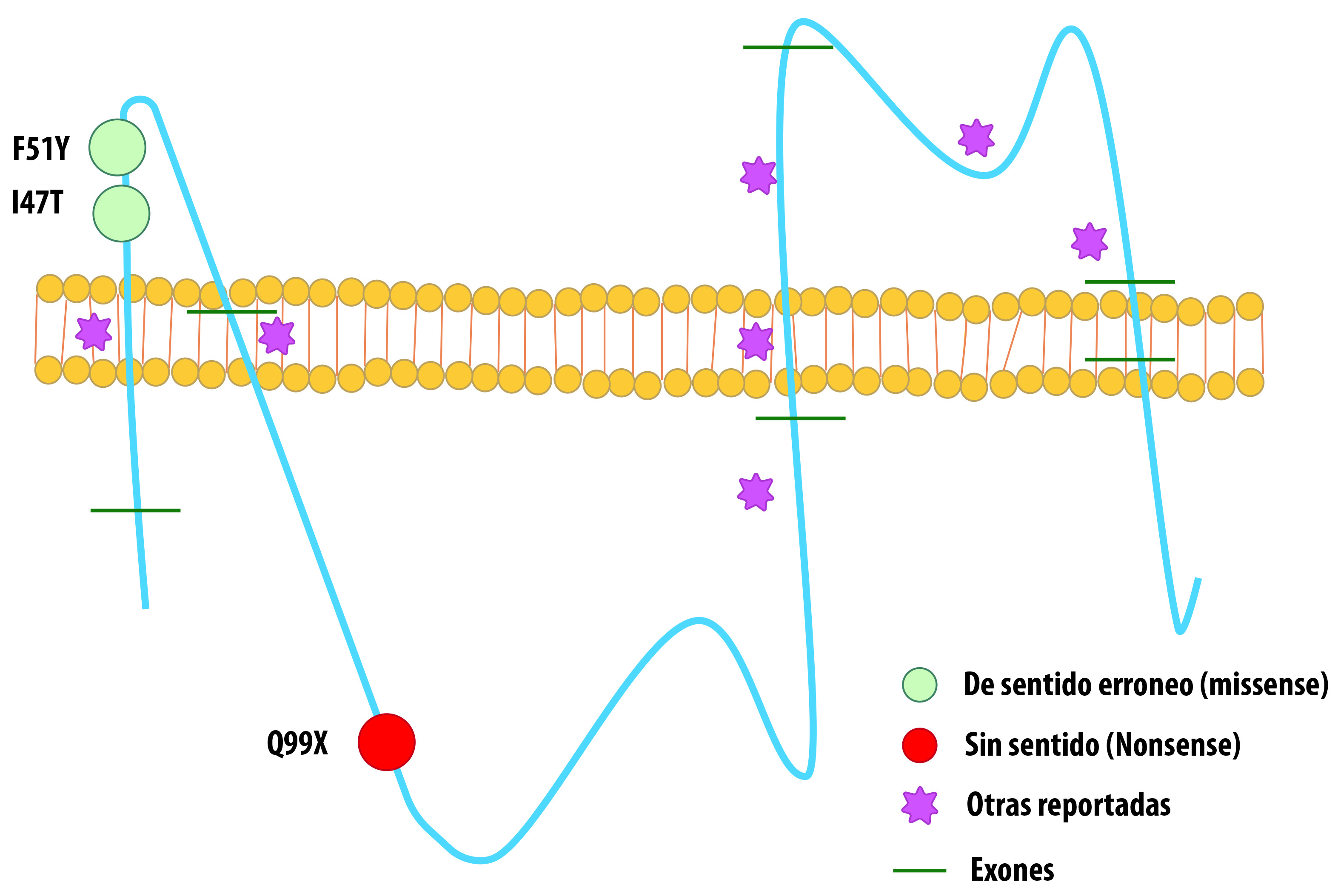
Figura 2 Mutaciones encontradas en nuestros pacientes y las previamente reportadas en PLP1. La figura recoge un esquema de la localización a nivel de la proteína de las mutaciones que se detectaron en los casos aquí presentados (Missense, Nonsense) vs. Las mutaciones reportadas previamente en la literatura (Otras reportadas).
El gen PLP1 está localizado en la región cromosómica Xq22, tiene una longitud de 17 kb, contiene 7 exones y 6 intrones. El exón 1 codifica solo para una metionina y los exones 2, 3, 4, y 5 codifican los dominios hidrofóbicos y la secuencia de cadenas hidrofílicas. El dominio C terminal transmembranal es codificado por los exones 6 y 7. Este último contiene una secuencia C terminal hidrofílica y la región 3’ no trascrita 14,15. El gen PLP1 Codifica para la proteína proteolipídica de mielina tipo 1 llamada también lipofilina (PLP1) de 276 aminoácidos y su isoforma generada por splicing alternativo DM20. Tanto PLP1 como DM20 son proteínas de membrana muy hidrofóbicas. La proteína DM20 pierde 35 residuos dentro de su loop intracelular y tanto PLP/DM20 constituyen más del 50% de las proteínas de la mielina compacta en el sistema nervioso central del adulto 16. Las funciones de PLP/DM20 no han sido descritas totalmente; sin embargo, es claro que son necesarias para el ensamblaje y estabilidad física de la mielina del sistema nervioso central. No sobra decir que las mutaciones en el gen PLP1 han sido ampliamente relacionadas como causa de PMD y SPG2. Los estudios en pacientes con estas patologías, así como modelos en ratón, han permitido definir qué PLP/DM20 participa en la formación de la línea intra-período de mielina, la compactación de la mielina, la adhesión de las vainas de mielina a la membrana del oligodendrocito y el recubrimiento de mielina del axón. Un amplio tipo de mutaciones ha sido descrito en este grupo de individuos, siendo la forma más frecuentemente reportada la duplicación de gen PLP117,18,19.
La mutación c.140T>C que encontramos en dos de nuestros pacientes con fenotipo connatal fue recientemente reportada por Hoffman y col. pero en PMD clásica 5. Grossi et al.20,. reportaron una mutación similar en un fenotipo clásico, la microduplicación en el exon 2 de PLP (c.134_140dup7) con corrimiento en el marco en lectura (p.Ile47IlefsX4) y repercusión en una proteína truncada en 4 aminoácidos corriente abajo 21. Será importante en el futuro establecer la funcionalidad bioquímica de la posición Isoleucina 47 con respecto a su impacto en el fenotipo connatal, ya hasta el momento no existen estudios funcionales que permitan comprobar el efecto in vivo de ella.
Las mutaciones nonsense c.609C>T (p.Q99X) y missense y c.152T>A (p.F51Y) no han sido reportadas previamente como causales, pero la predicción por SIFT y PolyPhen de su efecto en la proteína sugiere que sean probablemente deletéreas (Scores de 0.82 y 0.98 en Polyphen respectivamente). Estas mutaciones comprometen dos dominios funcionales de la proteína, en el caso de la c.609C>T (p.Q99X) se ve afectado el dominio citoplasmático y en el caso de la c.152T>A (p.F51Y) un dominio topológico extracelular 14,16.
En un menor porcentaje de casos se ha reportado que pueden ocurrir triplicaciones, triplicaciones parciales e incluso quintuplicaciones del gen PLP1, y en menos del 2% de los casos se presentan deleciones completas o parciales de este gen 22,23,24.
Conclusiones
En nuestro conocimiento, este es el primer reporte latinoamericano de una serie de pacientes con PMD en el que se encuentra una mayor frecuencia de afectados con PMD clásica con puntajes para discapacidad moderada en quienes se detectaron más mutaciones puntuales que duplicaciones del gen PLP1 como la causa. En los dos pacientes con la forma connatal de la enfermedad que presentamos, a pesar de realizar un diagnóstico más temprano, la discapacidad fue severa y el pronóstico vital fue siempre pobre.
Consideramos que, en esta población, se debería contemplar un algoritmo diagnóstico que, tras la alta sospecha clínica y el soporte con neuroimagen y otros paraclínicos, confirme en simultáneo la alteración molecular de rearreglos tipo delecion / duplicación y la presencia de mutaciones puntuales en el gen PLP1, mediante secuenciación Sanger asociada al análisis de la dosis génica por MLPA o por otra técnica molecular disponible para ese propósito.
A pesar de su carácter discapacitante, los pacientes con formas leves o moderadas de la Enfermedad de Pelizaeus Merzbacher tienen una esperanza de vida larga, con escasos reportes de enfermedad grave que causa el fallecimiento en la segunda década de la vida y la enfermedad en ellos progresa lentamente tras la adolescencia. Es importante mejorar el algoritmo diagnóstico para disminuir los tiempos antes del establecimiento completo de los tratamientos paliativos y para el adecuado asesoramiento genético de las familias, además que, de tratarse de la forma connatal, la muerte del paciente ocurrirá generalmente en la primera década de la vida, por lo que es importante el diagnóstico previo.
REFERENCIAS
1. Pelizaeus F. Über eine eigentümliche Form spastischer Lähmung mit cerebral Erscheinungen auf hereditärer Grund- lage. Arch Psychiatr Nervenkr. 1885; 16: 698 - 710. [ Links ]
2. Merzbacher L. Eine eigenartige familiäre-hereditäre Erkrankungsform (Aplasia axialis extracorticalis congenita). Z Ges Neurol Psychiatr. 1910; 3: 1 - 138. [ Links ]
3. Gencic S, Abuelo D, Ambler M, Hudson LD. Pelizaeus - Merzbacher Disease: An X - linked Neurologic Disorder of Myelin Metabolism with a Novel Mutation in the Gene Encoding Proteolipid Protein. Am J Hum Genet. 1989; 45: 435 - 442. [ Links ]
4. Genetics Home Reference. Gene PLP1. Fecha de Consulta: Agosto 01 de 2015. NIH: USA. 2008. Disponible en: http://ghr.nlm.nih.gov/gene/PLP1. [ Links ]
5. Hofman-Zacharska D, Mierzewska H, Szczepanik E, Poznanski J, Mazurczak T, Jakubiuk-Tomaszuk A, et al. The espectrum of PLP1 gene mutations in patients with classical form of the Pelizaeus Merzbacher disease. Developmental Period Medicine. 2013; XVII(4): 293 - 300. [ Links ]
6. Hobson GM, Garbern JY. Pelizaeus-Merzbacher disease, Pelizaeus-Merzbacher-like disease 1, and related hypomyelinating disorders. Semin Neurol. 2012; 32(1): 62 - 67. [ Links ]
7. Lee E, Yum M, Choi H, Yoo H, You SJ, Lee EH, et al. Magnetic resonance imaging and spectroscopic analysis in 5 cases of Pelizaeus-Merzbacher disease: metabolic abnormalities as diagnostic tools. Korean J Pediatr. 2012; 55(10): 397-402. [ Links ]
8. Inoue K. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics. 2005; 6: 1 - 16. [ Links ]
9. Hobson GM, Kamholz J. PLP1-Related Disorders. In: Adam MP, Ardinger HH, Pagon RA, et al. Gene Reviews. Seattle (WA): University of Washington, Seattle; 2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1182/ [ Links ]
10. Boulloche J, Aicardi J. Pelizaeus-Merzbacher disease: clinical and nosological study. J Child Neurol. 1986;1:233-239. [ Links ]
11. Bonkowsky JL, Nelson C, Kingston JL, Filloux FM, Mundorff MB, Srivastava R. The burden of inherited leukodystrophies in children. Neurology. 2010;75:718-725. [ Links ]
12. Garbern JY, Yool DA, Moore GJ, Wilds IB, Faulk MW, Klugmann M, et al. Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain. 2002;125:551-561. [ Links ]
13. Laukka JJ, Stanley JA, Garbern JY, Trepanier A, Hobson G, Lafleur T, et al. Neuroradiologic correlates of clinical disability and progression in the X-Linked leukodystrophy Pelizaeus-Merzbacher disease. J Neurol Sci. 2013; 335(0): 75-81. [ Links ]
14. Diehl HJ, Schaich M, Budzinski RM, Stoffel W. Individual exons encode the integral membrane domains of human myelin proteolipid protein. Proc Natl Acad Sci Proc U S A. 1986;83:9807-9811. [ Links ]
15. Yool DA, Edgar JM, Montague P, Malcolm S. The proteolipid protein gene and myelin disorders in man and animal models. Hum Molec Genet. 2000; 9:987-992. [ Links ]
16. Stoffel W, Hillen H, Giersiefen H. Structure and molecular arrangement of proteolipid protein of central nervous system myelin. Proc Natl Acad Sci U S A. 1984; 81: 5012-5016. [ Links ]
17. Hobson GM, Davis AP, Stowell NC, Kolodny EH, Sistermans EA, de Coo IFM, et al. Mutations in noncoding regions of the proteolipid protein gene in Pelizaeus-Merzbacher disease. Neurology. 2000;55:1089-1096. [ Links ]
18. Gow A, Lazzarin RA. A cellular mechanism governing the severity of Pelizaeus-Merzbacher disease. Nat Genet. 1996;13:422-428. [ Links ]
19. Cailloux F, Gauthier-Barichard F, Mimault C, Isabelle V, Courtois V, Giraud G, et al. Genotype-phenotype correlation in inherited brain myelination defects due to proteolipid protein gene mutations. Eur J Hum Genet. 2000;8:837-845. [ Links ]
20. Grossi S, Regis S, Biancheri R, Mort M, Lualdi S, Bertini E, et al. Molecular genetic analysis of the PLP1 gene in 38 families with PLP1-related disorders: identification and functional characterization of 11 novel PLP1 mutations. Orphanet J Rare Dis. 2011; 6:40. doi: 10.1186/1750-1172-6-40 [ Links ]
21. UniProt. KB - P60201 (MYPR_HUMAN). UniProt Consortium. Accessed: 03 August 2015. Available from: http://www.uniprot.org/uniprot/P60201. [ Links ]
22. Woodward K, Kendall E, Vetrie D. Variation in PLP gene duplications causing Pelizaeus-Merzbacher disease. Am J Hum Genet. 1998; 63:A394. [ Links ]
23. Combes P, Bonnet-Dupeyron MN, Gauthier-Barichard F, Schiffmann R, Bertini E, Rodriguez D, et al. PLP1 and GPM6B intragenic copy number analysis by MAPH in 262 patients with hypomyelinating leukodystrophies: identification of one partial triplication and two partial deletions of PLP1. Neurogenetics. 2006;7:31-7. [ Links ]
24. Wolf NI, Sistermans EA, Cundall M, Hobson GM, Davis-Williams AP, Palmer R, et al. Three or more copies of the proteolipid protein gene PLP1 cause severe Pelizaeus-Merzbacher disease. Brain. 2005;128:743-51. [ Links ]
Recibido: 09 de Septiembre de 2016; Revisado: 25 de Mayo de 2017; Aprobado: 02 de Noviembre de 2017
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
