










Serviços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkColombia Médica
versão On-line ISSN 1657-9534
Colomb. Med. vol.54 no.3 Cali jul./set. 2023 Epub 30-Set-2023
https://doi.org/10.25100/cm.v54i3.5667
Articulo original
Caracterización clínica y desenlaces de una cohorte de pacientes colombianos con amiloidosis AL
1
2
 http://orcid.org/0000-0002-8398-5807
http://orcid.org/0000-0002-8398-5807
3
4
http://orcid.org/0000-0002-5402-4599
5
http://orcid.org/0009-0003-0133-4088
2
6
http://orcid.org/0000-0001-6748-6971
2
6
http://orcid.org/0000-0001-8399-7253
1 Hospital San Vicente Fundación Rionegro , Departamento de Hematología, Rionegro, Colombia.
2 Universidad de Antioquia, Sección de Hematología Clínica, Departamento de Medicina interna, Facultad de Medicina, Medellín, Colombia.
3 Universidad de Antioquia, Facultad de Medicina Departamento de Medicina interna, Medellín, Colombia.
4 Hospital Alma Mater de Antioquia, Departamento de Medicina Interna, Medellín, Colombia.
5 Hospital Pablo Tobón Uribe; Medellín; Colombia.
6 Hospital San Vicente Fundación Medellín, Departamento de Hematología, Medellín, Colombia.
7 Hospital Pablo Tobón Uribe, Departamento de Hematología, Medellín, Colombia.
8 Universidad de Antioquia, Instituto de Investigaciones Médicas, Facultad de Medicina, Medellín, Colombia.
Antecedentes:
La amiloidosis por amiloide de cadenas ligeras (AL) se caracteriza por el depósito de fibrillas amiloides derivadas de cadenas ligeras de inmunoglobulinas monoclonales, lo que resulta en disfunción multiorgánica. Existen datos limitados sobre las características clínicas de la amiloidosis AL.
Objetivo:
Este estudio tiene como objetivo describir las características clínicas, tratamientos y desenlaces en pacientes colombianos con amiloidosis AL.
Métodos:
Se llevó a cabo un estudio descriptivo retrospectivo en tres centros de alta complejidad en Medellín, Colombia. Se incluyeron adultos con diagnóstico de amiloidosis AL entre 2012 y 2022. Se analizaron datos clínicos, de laboratorio, histológicos, de tratamiento y de supervivencia.
Resultados:
El estudio incluyó 63 pacientes. La afectación renal fue más prevalente (66%), seguida de la afectación cardíaca (61%). El 61% de los pacientes presentaron afectación multiorgánica. El depósito amiloide se detectó con mayor frecuencia en la biopsia renal (40%). El tratamiento basado en bortezomib se utilizó en el 68%, y el 23.8% recibió altas dosis de quimioterapia con trasplante autólogo de progenitores hematopoyéticos (ADQT-TAPH). Se observó respuesta hematológica en el 95% de los pacientes con datos disponibles. La respuesta de órgano cardíaca y renal fue del 15% y 14%, respectivamente. La mediana de la supervivencia global fue de 45.1 meses (IC del 95%: 22.2-63.8). En el análisis multivariado, la afectación cardíaca se asoció significativamente con una supervivencia global inferior (HR 3.27; IC del 95%: 1.23-8.73; p=0.018), ADQT-TAPH mostró una tendencia no significativa hacia una mejora en la supervivencia global (HR 0.25; IC 95%: 0.06-1.09; p=0.065).
Conclusiones:
En este estudio de pacientes colombianos con amiloidosis AL, la afectación renal fue más frecuente que la afectación cardíaca. La supervivencia global y la afectación multiorgánica fueron consistentes con datos de otras regiones del mundo. El análisis multivariado identificó la afectación cardíaca y ADQT-TAPH como posibles factores pronósticos.
Palabras clave: Amiloidosis; bortezomib; lenalidomida; trasplante; cadenas ligeras de inmunoglobulinas; daratumumab; proteínas amiloidogénicas; células plasmáticas; mieloma múltiple; respuesta al trasplante autólogo de progenitores hematopoyéticos
Background:
Amyloid light chain (AL) amyloidosis is characterized by amyloid fibril deposition derived from monoclonal immunoglobulin light chains, resulting in multiorgan dysfunction. Limited data exist on the clinical features of AL amyloidosis.
Objective:
This study aims to describe the clinical characteristics, treatments, and outcomes in Colombian patients with AL amyloidosis.
Methods:
A retrospective descriptive study was conducted at three high-complexity centers in Medellín, Colombia. Adults with AL amyloidosis diagnosed between 2012 and 2022 were included. Clinical, laboratory, histological, treatment, and survival data were analyzed.
Results:
The study included 63 patients. Renal involvement was most prevalent (66%), followed by cardiac involvement (61%). Multiorgan involvement occurred in 61% of patients. Amyloid deposition was most commonly detected in renal biopsy (40%). Bortezomib-based therapy was used in 68%, and 23.8% received high-dose chemotherapy with autologous hematopoietic stem cell transplantation (HDCT-ASCT). Hematological response was observed in 95% of patients with available data. Cardiac and renal organ responses were 15% and 14%, respectively. Median overall survival was 45.1 months (95% CI: 22.2-63.8). In multivariate analysis, cardiac involvement was significantly associated with inferior overall survival (HR 3.27; 95% CI: 1.23-8.73; p=0.018), HDCT-ASCT had a non-significant trend towards improved overall survival (HR 0.25; 95% CI: 0.06-1.09; p=0.065).
Conclusions:
In this study of Colombian patients with AL amyloidosis, renal involvement was more frequent than cardiac involvement. Overall survival and multiorgan involvement were consistent with data from other regions of the world. Multivariate analysis identified cardiac involvement and HDCT-AHCT as possible prognostic factors.
Keywords: Amyloidosis; bortezomib; lenalidomida; transplantation; immunoglobulin light-chain; daratumumab; amyloidogenic proteins; plasma cells; multiple myeloma; hematopoietic stem cell transplantation response
Contribución del estudio
| 1) ¿Por qué se realizó este estudio? |
| Describir las características clínicas, los patrones de tratamiento y los resultados de los pacientes con amiloidosis AL en la población colombiana a través de un análisis retrospectivo en tres centros altamente especializados de Medellín, Colombia |
| 2) ¿Cuáles fueron los resultados más relevantes del estudio? |
| Se halló una mayor frecuencia de afectación renal en comparación con la afectación cardiaca. Además, la frecuencia de afectación multiorgánica fue similar a la de otras regiones, como Europa y Asia. La tasa de supervivencia global observada en este estudio fue comparable a la comunicada en una publicación europea reciente. El análisis multivariante realizado en el estudio identificó la afectación cardiaca y la consolidación con HDCT-AHCT como factores pronósticos potenciales. |
| 3¿Qué aportan estos resultados? |
| Este estudio reporta las características y resultados de pacientes colombianos con amiloidosis AL, identifica factores pronósticos potenciales en nuestra población y proporciona una base para diseñar futuros estudios con un alcance más amplio. |
Introducción
La amiloidosis primaria o de cadenas ligeras (AL) es la forma más común de amiloidosis sistémica 1. Esta enfermedad se origina a partir de un clon de células plasmáticas o, con menos frecuencia, a partir de un linfoma de bajo grado, como la leucemia linfocítica crónica (LLC), el linfoma linfoplasmocítico o el linfoma de zona marginal, capaz de secretar cadenas ligeras de inmunoglobulina monoclonal con plegamiento inestable debido a mutaciones en su región variable 2. En el 80% de los casos, la cadena ligera comprometida es la cadena lambda debido a una predisposición amiloidogénica intrínseca más alta que la cadena kappa 3,4. Esta proteína amiloidogénica activa la formación, agregación y estabilización de fibrillas amiloides insolubles que presentan una alta afinidad por la tinción de rojo Congo, característica de esta enfermedad 3. La presencia de depósitos de amiloide en diferentes tejidos conduce a disfunción de órganos mediada por proteotoxicidad y reemplazo de la arquitectura típica, lo que finalmente resulta en signos y síntomas sistémicos 5,6.
La amiloidosis AL es una enfermedad rara y difícil de diagnosticar, lo que resulta en limitados datos publicados sobre su epidemiología global y características clínicas 7,8. Datos recientemente actualizados de una cohorte de pacientes en Olmsted, Minnesota, estiman una incidencia de 8.9 a 12 casos por millón de personas-año 9,10; datos de otras regiones del mundo estiman una incidencia de 3 a 12 casos por millón de personas-año 11-13. Una revisión sistemática de la literatura recientemente publicada utilizó datos publicados de América del Norte, América del Sur, Europa y Asia para evaluar la epidemiología global de la amiloidosis AL, estimando una incidencia y prevalencia a 20 años de 10.44 y 51 casos por millón de personas-año, respectivamente 14.
A excepción del sistema nervioso central, las cadenas ligeras de inmunoglobulina monoclonal pueden depositarse virtualmente en cualquier tejido, lo que conduce a una amplia gama de manifestaciones clínicas y afectación de órganos, incluyendo el corazón, riñones, hígado, tejidos blandos, sistema nervioso periférico, entre otros, los cuales son los principales marcadores pronósticos 8,15,16.
El tratamiento actual de la amiloidosis AL se enfoca principalmente en suprimir la neoplasia maligna subyacente para reducir la producción de cadenas ligeras amiloidogénicas 2. No existen terapias aprobadas para el tratamiento de la amiloidosis AL, de manera que el tratamiento para esta enfermedad ha sido heterogéneo, basado en combinaciones adaptadas de tratamientos para el mieloma múltiple o linfomas de bajo grado 17, incluyendo regímenes como MDex (melfalán y dexametasona), VMD (bortezomib, melfalán y dexametasona), CyBorD (ciclofosfamida, bortezomib y dexametasona) 18,19, y consolidación de la respuesta con quimioterapia de alta dosis seguida de trasplante autólogo de progenitores hematopoyéticos (QTAD-TAPH) en pacientes elegibles 20. Datos recientemente publicados demuestran resultados prometedores con la adición de daratumumab al régimen CyBorD, logrando tasas de respuesta y supervivencia libre de progresión mejores que CyBorD 21. El pronóstico de la amiloidosis AL está influenciado por múltiples factores, incluyendo la gravedad de la afectación cardíaca, que se ha establecido como el principal determinante de la supervivencia 2,22,23, la coexistencia con otras neoplasias como el mieloma múltiple 24, la profundidad de la respuesta hematológica 25, entre otros.
A pesar de que algunos estudios reportan datos de América Latina, hay escasa información sobre pacientes colombianos. Este estudio tiene como objetivo describir las características clínicas, patrones de tratamiento y desenlaces de pacientes con amiloidosis AL en la población colombiana a través de un análisis retrospectivo en tres centros altamente especializados en Medellín, Colombia.
Materiales y Métodos
Población y Muestra
La población del estudio consistió en pacientes mayores de 18 años, de ambos sexos, diagnosticados con amiloidosis AL, que habían sido atendidos como pacientes ambulatorios u hospitalizados por esta condición en el Hospital Universitario San Vicente Fundación de Medellín, el Hospital San Vicente Fundación de Rionegro o el Hospital Pablo Tobón Uribe entre enero de 2012 y diciembre de 2022.
Se incluyeron todos los pacientes que cumplían con los criterios de inclusión durante el período del estudio, excluyendo únicamente a aquellos que no tenían información sobre las variables de interés.
El diagnóstico de la amiloidosis AL requirió demostración histológica de depósito amiloide mediante tinción de rojo Congo positiva y birrefringencia verde manzana bajo luz polarizada, asociada a la presencia de gammapatía monoclonal identificada por inmunofijación en suero/orina, relación kappa/lambda anormal indicando un exceso de cadenas ligeras, o la presencia de células plasmáticas clonales en la biopsia de médula ósea 7,21. Otros tipos de amiloidosis (amiloidosis AA, amiloidosis por transtirretina, amiloidosis localizada, amiloidosis hereditaria, etc.) fueron excluidas. La clasificación de la afectación de órganos, la respuesta de órganos y la respuesta hematológica se definieron de acuerdo con los criterios del 10º Simposio Internacional sobre Amiloide y Amiloidosis publicados en 2005 26 y las guías para la realización y reporte de ensayos clínicos en amiloidosis sistémica de cadena ligera publicadas en 2012 27.
Variables
Se evaluaron variables demográficas, clínicas, de laboratorio, histológicas, estado funcional, tratamiento y supervivencia global. Se realizó un análisis univariado para determinar el efecto potencial de neoplasias asociadas, afectación renal, afectación cardíaca y QTDA-TAPH en la supervivencia global de la población evaluada, y se llevó a cabo un análisis multivariado para ajustar posibles variables de confusoras. La información clínica se obtuvo retrospectivamente de los registros médicos electrónicos y fue revisada de forma independiente por seis médicos.
Análisis Estadístico
Para las variables categóricas, se utilizaron distribuciones de frecuencia absoluta y relativa para cada categoría. Para las variables numéricas, se utilizaron medidas de tendencia central como la media con desviación estándar o la mediana con rango intercuartílico.
La supervivencia global se analizó utilizando el modelo de Kaplan-Meier con mediana de supervivencia e intervalos de confianza del 95% (IC). El análisis de regresión de Cox evaluó los factores asociados, verificando el supuesto de proporcionalidad de riesgos. Se estimaron Hazard Ratios (HR) observados y ajustados junto con IC del 95% y valores de p. Se incluyeron en el ajuste las variables con p <0.25 en el análisis multivariado. A través del análisis post hoc, se estableció, con un alfa de 0.05, que la potencia estadística es mayor o igual a 0.80 para HR ≥2.7 o HR: ≤0.31.
Consideraciones éticas
La Dirección de Investigación y el Comité de Ética del Hospital San Vicente Fundación de Medellín, el Hospital San Vicente Fundación de Rionegro y el Hospital Pablo Tobón Uribe aprobaron este estudio. Se garantizó la confidencialidad de los datos para prevenir la identificación de pacientes durante las etapas de publicación.
Resultados
Características de los pacientes
Este estudio incluyeron 63 pacientes con amiloidosis AL, se evaluaron características demográficas, clínicas, histológicas, de tratamiento y desenlaces de supervivencia. La Tabla 1 proporciona detalles de las características clínicas y de laboratorio analizadas.
Tabla 1 Características Demográficas y Clínicas de los Pacientes con Amiloidosis AL
| Characteristics | Number of patients (%) |
|---|---|
| Sexo | |
| Masculino | 36 (57) |
| Edad (años) | |
| Media (rango) | 60 (34-87) |
| ECOG | |
| 0 | 4 (6.0) |
| 1 | 25 (40) |
| 2 | 14 (22) |
| 3 | 7 (11) |
| sin dato | 13 (21) |
| Trombocitosis (PLTs >450,000/mm3) | 6 (9.5) |
| Anemia (Hb <10 g/dL) | 16 (25) |
| Creatinina > 1.5 mg/dL | 9 (14)a |
| Promedio de TFG por CKD-EPI (ml/min/1.73 m²) | 73 (DS: 29.5)a |
| TFG <50 ml/min/1.73 m² | 11 (22.9)a |
| Cadena ligera involucrada | |
| Lambda | 45 (71) |
| Kappa | 13 (21) |
| Sin dato | 5 (8.0) |
| Cadena pesada involucrada | |
| IgG | 19 (30.0) |
| IgA | 9 (14.0) |
| IgM | 2 (3.0) |
| IgD | 1 (1.5) |
| Sin cadena pesada involucrada | 27 (42.0) |
| Sin dato | 6 (9.5) |
| Neoplasia Asociada | |
| Mieloma múltiple | 37 (58.0) |
| Leucemia linfocítica crónica | 1 (1.5) |
| Ninguna | 25 (40.0) |
| Promedio de infiltración por células plasmáticas en médula ósea | 19% (DS: 22.3) |
| Infiltración de células plasmáticas en médula ósea ≥10% | 33 (52) |
| Relación kappa/lambda anormal (<0.26 or >1.65) | 29 (87)b |
| Promedio de dFLC al ingreso (mg/L) | 9,553 (DS: 17,519) |
| Promedio de proteinuria al ingreso (mg/24h) | 5,444 (DS: 6,334)c |
| Proteinuria en rango nefrótico (≥3,500 mg/24 hours) | 22 (49%) |
| Promedio de BNP al ingreso (ng/L) | 663 (DS: 619)d |
| BNP >81 ng/L | 20 (86%) |
| BNP >700 ng/L | 10 (43%) |
| Afectación de órgano | |
| Renal | 42 (66.0) |
| Cardiaco | 39 (61.0) |
| Sistema nervioso periférico | 22 (34.0) |
| Tejidos blandos | 17 (26.0) |
| Hepático | 16 (25.0) |
| Pulmonar | 8 (12.6) |
| Gastrointestinal | 6 (9.5) |
| Dos o más órganos | 39 (61) |
TFG = tasa de filtración glomerular. dFLC = diferencia de cadena ligera involucrada-no involicrada. DS= Desviación estándar. aInformación disponible en 48 patients. bInformación disponible en 33 patients. cInformación disponible en 45 patients. dInformación disponible en 23 patients.
La edad promedio de los pacientes fue de 60 años (DE: 14.2), con un rango de 34 a 87 años. Treinta y seis (57%) pacientes eran hombres. Cuarenta y tres (68%) pacientes tenían un estado funcional ECOG ≤2. De acuerdo con los criterios del Simposio Internacional sobre Amiloide y Amiloidosis 26, 42 (66%) pacientes tenían afectación renal, seguido por afectación cardíaca (61%), afectación del sistema nervioso periférico (34%), afectación de tejidos blandos (26%), afectación hepática (25%), afectación pulmonar (12.6%) y afectación gastrointestinal (9.5%). La mayoría de los pacientes (61%) tenían afectación de dos o más órganos. Entre los pacientes con afectación renal, 22 (52%) presentaron proteinuria en rango nefrótico (≥3500 mg/24 horas), lo que representa el 34% de todos los pacientes evaluados. Diez (43%) de los pacientes con afectación cardíaca tenían niveles de péptido natriurético cerebral (BNP) ≥700 ng/L.
Hallazgos histológicos y de laboratorio
Se obtuvo información sobre los resultados histológicos de 57 (90%) pacientes, incluyendo 63 biopsias tomadas de diferentes tejidos. Entre los pacientes analizados con resultados histológicos disponibles, el depósito amiloide se detectó con mayor frecuencia en biopsias renales (40%), seguido de biopsias de grasa abdominal (17%), biopsias de médula ósea (12%), miocardio (8%), hígado (8%), ganglio linfático (5%), tracto gastrointestinal (5%), piel (5%), glándula salival menor (3%) y pulmón (3%).
En cuanto a los resultados de laboratorio durante el abordaje diagnóstico inicial, 16 (25%) pacientes tenían anemia, 6 (9.5%) tenían trombocitosis, nueve pacientes (14%) presentaron creatinina mayor a 1.5 mg/dL y 11 pacientes (22.9%) tenían una tasa de filtración glomerular (TFG) < 50 ml/min/1.73 m² en el momento del diagnóstico. En la mayoría de los casos, la cadena ligera de inmunoglobulina involucrada fue la cadena lambda (71%), mientras que el isotipo kappa estaba presente en 13 pacientes (20%). De los 33 pacientes con información sobre la concentración de cadenas ligeras libres en suero, 29 (87%) tenían una relación kappa/lambda anormal. La distribución de las cadenas pesadas de inmunoglobulina fue la siguiente: la inmunofijación en suero por electroforesis fue positiva en 58 pacientes (90%), incluyendo 19 (30%) pacientes con IgG, 9 (14%) con IgA, 2 (3%) con IgM, 1 (1.5%) con IgD y 27 (42%) pacientes con compromiso aislado de cadenas ligeras de inmunoglobulina.
Treinta y ocho pacientes (60%) presentaron una neoplasia asociada a la amiloidosis AL, 37 (58.5%) tenían mieloma múltiple y 1 (1.5%) tenía leucemia linfocítica crónica. El porcentaje promedio de infiltración de células plasmáticas en la médula ósea fue del 19% (DE: 22.3), y 33 (52%) pacientes presentaban infiltración de la médula ósea del 10% o más por células plasmáticas.
Tratamiento, respuesta y supervivencia
La población del estudio recibió tratamiento en diferentes centros de atención médica durante diez años, utilizando varios regímenes de tratamiento, detallados en la Tabla 2. Las terapias basadas en bortezomib (VCD, VD y Dara-VCD) fueron las más comúnmente utilizadas (68%), seguidas por combinaciones con medicamentos inmunomoduladores como talidomida o lenalidomida (14%), y terapias basadas en agentes alquilantes como melfalán o ciclofosfamida (9%); cinco (8%) pacientes no recibieron terapia de inducción, y 15 (23.8%) pacientes recibieron QTAD-TAPH después de la terapia de inducción.
Tabla 2 Regímenes de Tratamiento de Primera Línea y Consolidación con QTAD-TAPH para Pacientes con Amiloidosis AL
| Régimen | Numero de pacientes (%) |
|---|---|
| VCD, bortezomib, ciclofosfamida y dexametasona | 37 (59) |
| VD, bortezomib y dexametasona | 5 (8.0) |
| Daratumumab-VCD, daratumumab + bortezomib, ciclofosfamida y dexametasona | 1 (1.6) |
| DRD, daratumumab, lenalidomida y dexametasona | 1 (1.6) |
| VRD, bortezomib, lenalidomida y dexametasonea | 2 (3.1) |
| VTD, bortezomib, thalidomide, and dexamethasone | 1 (1.6) |
| MDT, melfalán, Dexametasona y talidomida | 3 (4.7) |
| CTD, ciclofosfamida, talidomida y dexametasona | 2 (3.1) |
| Mdex, melfalán y dexametasona | 2 (3.1) |
| MDV, melfalán, Dexametasona y vincristina | 1 (1.6) |
| CD, ciclofosfamida y dexametasona | 3 (4.7) |
| None | 5 (8.0) |
| QTAD-TAPH, quimioterapia de altas dosis seguida de transplante autólogo de progenitores hematopoyéticos. | 15 (23.8) |
Se obtuvo información sobre la respuesta hematológica de 21 pacientes. La respuesta hematológica se logró en 20 pacientes con el tratamiento de primera línea, de los cuales 10 lograron una respuesta hematológica completa, 3 lograron una respuesta parcial muy buena, 7 lograron una respuesta parcial y 1 paciente experimentó progresión durante el tratamiento. La información sobre la respuesta hematológica no estaba disponible para 42 (66%) pacientes. Se observó respuesta de órganos en 6 (15%) de 39 pacientes con afectación cardíaca, 6 (14%) de 42 pacientes con afectación renal y 1 (6%) de 16 pacientes con afectación hepática.
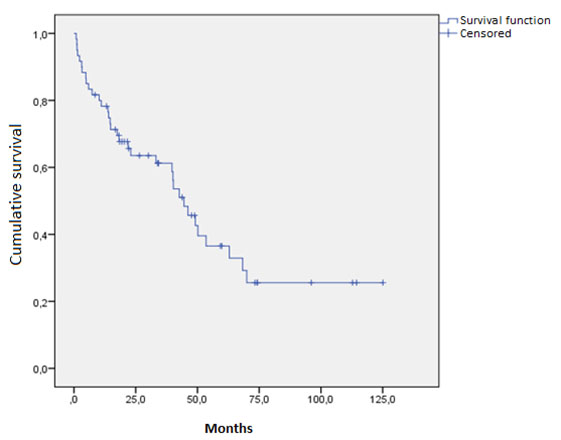
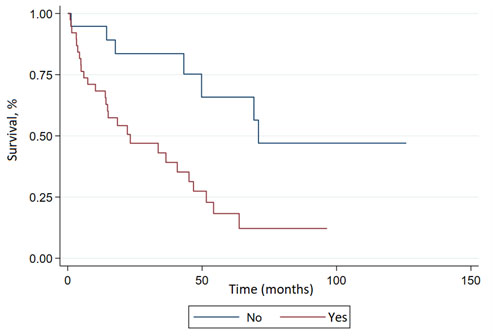
La mediana de supervivencia global de la población estudiada fue de 45.1 meses (IC del 95%: 22.2-63.8) (Figura 1), con un seguimiento promedio de 24.7 meses en la última consulta el 9 de marzo de 2023. El efecto de varios factores analizados en la supervivencia se muestra en la Tabla 3. La afectación cardíaca estuvo significativamente asociada con una supervivencia global inferior (HR: 3.27; IC del 95%: 1.23-8.73; p= 0.018) (Figura 2), mientras que la afectación renal (HR: 1.84; IC del 95%: 0.65-5.19; p= 0.248) o la presencia de una neoplasia asociada (HR: 0.98; IC del 95%: 0.49-1.97; p= 0.956) no tuvieron un impacto significativo en el pronóstico de la población evaluada. Los pacientes que recibieron QTAD-TAPH mostraron una tendencia no significativa (HR: 0.25; IC del 95%: 0.06-1.09; p= 0.065) hacia una mejora en la supervivencia global.
Tabla 3 Efecto de las Características Clínicas en la Supervivencia Global
| Características | Univariado | Multivariado | ||||||
|---|---|---|---|---|---|---|---|---|
| HR | Valor p | IC95% | HR | Valor p | IC95% | |||
| Afectación cardíaca | ||||||||
| Si | 3.47 | 0.004 | 1.48 | 8.14 | 3.27 | 0.018 | 1.23 | 8.73 |
| No | 1.00 | 1 | ||||||
| Afectación renal | ||||||||
| Si | 1.05 | 0.927 | 0.40 | 2.77 | 1.84 | 0.248 | 0.65 | 5.19 |
| No | 1.00 | 1 | ||||||
| Neoplasia asociada | ||||||||
| Si | 0.98 | 0.956 | 0.49 | 1.97 | ||||
| No | 1 | |||||||
| Sexo | ||||||||
| Masculino | 1.04 | 0.906 | 0.53 | 2.05 | ||||
| Femenino | 1.00 | |||||||
| Edad al diagnóstico | 1.01 | 0.685 | 0.98 | 1.04 | ||||
| Intervención | ||||||||
| QTAD-TAPH | ||||||||
| Si | 0.15 | 0.0114 | 0.04 | 0.65 | 0.25 | 0.065 | 0.06 | 1.09 |
| No | 1.00 | 1 | ||||||
Mediana de supervivencia global: 45.1 meses (95% CI: 22.2-63.8). Abreviaciones: QTAD-TAPH = quimioterapia de altas dosis seguida de trasplante autólogo de progenitores hematopoyéticos.
Discusión
Se realizó un análisis retrospectivo en pacientes con amiloidosis AL tratados en diferentes centros de Medellín, Colombia, para definir sus características y desenlaces, con el fin de enriquecer la información disponible sobre la enfermedad en nuestra región. La edad promedio al momento del diagnóstico en la población estudiada fue de 60 años, similar a lo observado en cohortes latinoamericanas 28,29, población de Norteamérica 30 y datos recientes de países europeos 14,17, entre otras regiones del mundo 30,31.
La amiloidosis AL puede estar asociada con otras neoplasias derivadas de células plasmáticas o linfocitos B 2; en nuestro estudio, la mayoría de los pacientes (58%) tenía mieloma múltiple asociado con amiloidosis AL, una proporción más alta en comparación con los resultados reportados por Kourelis et al. 24, quienes observaron una frecuencia de mieloma múltiple del 46% en su cohorte de pacientes con amiloidosis AL. En el estudio mencionado, los pacientes con amiloidosis AL y mieloma múltiple tenían una menor supervivencia global que los pacientes con amiloidosis AL sin neoplasia asociada, sin embargo, este efecto en el pronóstico no se observó en el análisis univariado de nuestro estudio.
La afectación orgánica, particularmente la afectación cardíaca y su gravedad, son los principales determinantes del pronóstico en pacientes con amiloidosis AL 2,23. La frecuencia de afectación multiorgánica en los pacientes analizados fue del 61%, similar a los datos recientemente publicados de una cohorte europea por Palladini et al. 17, donde el 63% de los pacientes diagnosticados con amiloidosis AL después de 2010 tenían afectación de 2 o más órganos. La frecuencia de afectación multiorgánica en nuestro estudio fue mayor que los datos publicados por Muchtar et al. 32, en pacientes norteamericanos, ya que su estudio consideró afectación multiorgánica a partir de tres o más órganos.
Respecto a la frecuencia individual de afectación de órganos, la mayoría de nuestros pacientes (66%) tenía afectación renal, seguido de cerca por la afectación cardíaca (61%); la información publicada sobre cuál de los dos órganos se ve más afectado en la amiloidosis AL es heterogénea. En un estudio publicado por Michael et al. (30), realizado en múltiples hospitales en Grecia, el 71% de los pacientes tenía afectación renal, seguido del 59% de los pacientes con afectación cardíaca. Estudios recientes en población china también muestran una frecuencia más alta de afectación renal que de afectación cardíaca 31. Por otro lado, la cohorte europea mencionada 17 y los resultados del estudio de Muchatr et al. 32, informaron hallazgos contrarios donde la afectación cardíaca fue más frecuente que la afectación renal. El estudio recientemente publicado por Posadas-Martínez et al. 28, en pacientes argentinos, también mostró una frecuencia más alta de afectación cardíaca. Aunque estas diferencias probablemente se deban a diferencias raciales 33, es importante tener en cuenta que en nuestro entorno, los biomarcadores de daño cardíaco como BNP y el péptido natriurético cerebral pro-B tipo natriurético (NT-proBNP) han estado disponibles por poco tiempo y en pocos centros de atención médica; por lo tanto, las diferencias en la frecuencia de afectación cardíaca y renal en comparación con otras cohortes también pueden explicarse por el subdiagnóstico de la afectación cardíaca, que incluye criterios como BNP elevado o NT-proBNP.
Aunque los sistemas de estadificación para la amiloidosis AL están disponibles desde 2004, se basan principalmente en la afectación cardíaca 2,23,34-36, utilizando marcadores de daño miocárdico como BNP, NT-proBNP, troponina cardíaca T (cTNT), troponina cardíaca T de alta sensibilidad (hs-cTNT) o troponina cardíaca I (cTnI). Ninguno de los pacientes analizados tenía estudios de troponina validados acorde a los sistemas de estadificación de la amiloidosis AL; solo se disponía de información sobre los niveles de troponina cardíaca I de alta sensibilidad (hs-cTnI), y como actualmente no existen escalas de estadificación publicadas que incorporen hs-cTnI, no fue posible realizar la estadificación de la enfermedad en nuestra población. Entre los sistemas de estadificación para la amiloidosis AL, la escala de Boston 34 incluye niveles de BNP sérico >700 ng/L como marcador de mal pronóstico. De los 23 (36%) pacientes en nuestro estudio que tenían niveles de BNP sérico al diagnóstico, el 43% tenía niveles de BNP >700 ng/L. Estudios futuros deberían explorar el impacto de este marcador en los desenlaces de pacientes con amiloidosis AL en Colombia.
El diagnóstico de la amiloidosis AL es histológico y, por lo tanto, siempre requiere la demostración de depósito amiloide mediante análisis histopatológico del tejido 2,37. Si bien las biopsias de grasa abdominal y glándula salival menor son alternativas mínimamente invasivas con sensibilidad adecuada 37,38, en los pacientes analizados, solo el 20% tenía tinción positiva para Congo rojo en uno de estos tejidos. En comparación, el depósito de amiloide se demostró en biopsias renales en el 40% de los casos. Esto enfatiza que los resultados negativos en biopsias de grasa abdominal o glándula salival menor no excluyen el diagnóstico de amiloidosis AL, y es necesario realizar biopsias del órgano afectado como parte del enfoque diagnóstico en pacientes sospechosos de tener esta enfermedad.
Desde la década de 2010, los protocolos de tratamiento basados en bortezomib se han convertido en los regímenes más utilizados en pacientes con amiloidosis AL 2,17. Esta tendencia también se observó en nuestra población de estudio, donde los regímenes basados en bortezomib fueron la terapia preferida en el 68% de los pacientes, logrando una respuesta hematológica parcial o mejor en 20 (98%) de los 21 pacientes con información disponible sobre la respuesta hematológica. La consolidación de la respuesta con QTAD-TAPH ha sido reportada como una estrategia superior a utilizar solo quimioterapia de inducción 17,20. De los pacientes evaluados, el 23.8% se consolidó con QTAD-TAPH, una proporción más alta que en estudios similares 17,31,32) donde el 4.5% al 11.4% de los pacientes recibieron QTAD-TAPH después de la terapia de inducción.
La población evaluada tuvo una mediana de supervivencia de 45.1 meses (IC del 95%: 22.2-63.8), similar a los datos reportados en estudios previos realizados en otras regiones, donde la supervivencia media variaba de 34 a 48 meses 17,30-32. En el análisis multivariado, los pacientes con afectación cardíaca tuvieron una menor supervivencia, sin diferencias en la supervivencia de los pacientes con afectación renal, un hallazgo similar a estudios previos como el trabajo publicado por Sidana et al. 22, donde la mediana de supervivencia de los pacientes con afectación renal fue mayor en comparación con los pacientes con afectación cardíaca o afectación de ambos órganos. El análisis multivariado también mostró una tendencia no significativa hacia una mayor supervivencia en los pacientes consolidados con QTAD-TAPH, resultados similares a los informados por Gerts et al. 20, quienes reportaron una mayor supervivencia a los 3 años en pacientes tratados con QTAD-TAPH después de la terapia de inducción en comparación con pacientes tratados solo con melfalán y dexametasona. Sin embargo, estudios recientes sugieren que la terapia secuencial con QTAD-TAPH reservada solo para pacientes con mala respuesta a regímenes basados en bortezomib podría lograr resultados similares 39. La Tabla 4 resume las características del presente estudio y otros estudios similares previamente realizados.
Tabla 4 Características de los estudios comparados.
| Región | Colombia | Grecia | China | Estados Unidos | Argentina | Europa (post-2010) |
|---|---|---|---|---|---|---|
| Año de publicación | 2024 | 2010 | 2016 | 2019 | 2022 | 2023 |
| Método de diagnóstico | Rojo Congo + Gammapatía monoclonal | Rojo Congo + Gammapatía monoclonal | Rojo Congo + Gammapatía monoclonal | Espectrometría de masas | ||
| Número de pacientes | 63 | 112 | 123 | 592 | 90 | 3,065 |
| Edad (rango) | 60 (34-87) | 62 (38-84) | 54 (34-82) | 63 (56-71) | 63 (50-76) | 64 (29-91) |
| Sexo | Masculino (57%) | Femenino (53%) | Masculino (67%) | Masculino (64%) | Masculino (54%) | Masculino (58%) |
| Anemia/Trombocitosis | 34.5% | |||||
| Función renal | ||||||
| Creatinina ≥ 1.5 | 14% | 25% | 28% (>1.2) | |||
| Promedio de TFG | 73 (DS: 29.5) | 60 +/- 51 | ||||
| TFG <50 ml/min/1.73 m² | 23% | 27% | ||||
| Proteinuria ≥3.5 g/24h | 49% | 39% | 65% | 35% (>2 g/24h) | ||
| Gammapatía monoclonal | ||||||
| Cadena ligera involucrada | Lambda (71%) | Lambda (86%) | Lambda (75%) | Lambda (71%) | ||
| Cadena pesada involucrada | IgG (30%) | IgG (41%) | IgG (33%) | |||
| Neoplasia asociada | MM (58%) | Excluido MM sintomático | Excluido MM | Incluido MM (NR) | ||
| Infiltración por CP > 10% | 52% | 23% (>30) | 13.8% (>5) | 54% | 54% | |
| Afectación de órgano | ||||||
| BNP >81 ng/L | 86% | |||||
| BNP >700 ng/L | 43% | |||||
| Renal | 66% | 71% | 98% | 53% | 68% | 66% |
| Cardíaca | 61% | 59% | 55% | 76% | 72% | 69% |
| SNP | 34% | 38% | 9.8% | 24% | 35% | 14% |
| Tejidos blandos | 26% | 31% | 18% | 12% | 20% | |
| Hepática | 25% | 18% | 13% | 18% | 16% | 13% |
| Pulmonar | 12.6% | 3.6% | 0.9% | |||
| GI | 9.5% | 16% | 72% | 17% | 36% | 7% |
| 2 o más | 61% | 91% | 25% (≥3) | 63% | ||
| Sitio de biopsia | Renal (40%) | Renal (86%) | ||||
| Tratamiento | ||||||
| Primera línea de tratamiento | Basado en bortezomib (68%) | VAD o altas dosis de esteroides (43%) | Basado en altas dosis de esteroide (34%) | Basado en bortezomib (30%) | Basado en bortezomib (75%) | Basado en bortezomib (74%) |
| QTAD-TAPH | 23.8% | 4.5% | 12.0% | 31.0% | 11.4% | 9.8% |
| Respuesta | ||||||
| Cardíaca | 15% | 6% | 4% | |||
| Renal | 14% | 26% | 84% | |||
| Hepática | 6% | 33% | 8% | |||
| Hematológica | 95% (HCR 47%) | 50% (HCR 14%) | 100% (HCR 44%) | 84% (HCR 34%) | 61% (HCR 47%) | 63% (HCR 23%) |
| Análisis de supervivencia | ||||||
| Mediana de supervivencia (meses) | 45 | 34 | 38 | 43 | 60 | 46 |
| Análisis multivariado | Afectación cardíaca | Afectación cardíaca. CP >30%. Edad >65. Cr >1.5. | Afectación cardíaca. Afectación renal. | Afectación cardíaca. Proteinuria >5 g/24 h. Edad >65. | Respuesta hematológica complete. | |
Cr= creatinina. GI= gastrointestinal. HCR= respuesta hematológica completa. IMiD= inmunomoduladores. MM= myeloma múltiple. NR= no reportado. CP= célula plasmática. SNP= sistema nervioso periférico. VAD= vincristina, doxorubicina, dexametasona.
Este estudio presenta varias limitaciones. En primer lugar, debido a su diseño retrospectivo, no se obtuvo información completa sobre algunas variables, como los niveles de biomarcadores de daño cardíaco, la evaluación de respuesta hematológica y la respuesta de órgano. A pesar de que la amiloidosis AL es una enfermedad rara y difícil de diagnosticar, el pequeño tamaño de muestra de nuestro estudio es una limitación adicional que podría haber influido en la ausencia de asociaciones estadísticas para algunas de las variables evaluadas en el análisis de supervivencia. Esto también impide estimar modelos multivariados con múltiples factores simultáneamente. En consecuencia, las asociaciones presentadas en este estudio son exploratorias y requieren confirmación en futuros estudios. Los métodos utilizados para el diagnóstico de la amiloidosis AL en nuestro estudio representan otra limitación, considerando las tasas de falsos positivos y falsos negativos de la tinción con rojo Congo, así como la presencia simultánea de gammapatía monoclonal en pacientes con amiloidosis transtirretina o amiloidosis hereditaria. La espectrometría de masas es el estándar de oro para caracterizar la proteína amiloide; sin embargo, no está disponible en nuestro país. No obstante, a nuestro conocimiento, este es el primer estudio que informa las características y desenlaces de pacientes colombianos con amiloidosis AL, proporcionando una base para el diseño de futuros estudios con un alcance más amplio.
Conclusión
Presentamos el primer estudio reportando las características y desenlaces de pacientes colombianos con amiloidosis AL. El grupo de pacientes analizados tenía una distribución de edad similar a la reportada en estudios previos, con una mayor frecuencia de afectación renal en comparación con la afectación cardíaca y una mayor frecuencia de afectación multiorgánica en comparación con población norteamericana, aunque similar a otras regiones del mundo, como Europa y Asia. La proporción de pacientes consolidados con QTAD-TAPH fue mayor que la reportada en otros estudios, con una tasa de supervivencia global similar a la observada en otras regiones del mundo. El análisis multivariado realizado en el estudio identificó la afectación cardíaca y la consolidación con QTAD-TAPH como posibles factores pronósticos.
References
1. Dasari S, Theis JD, Vrana JA, Rech KL, Dao LN, Howard MT, et al. Amyloid typing by mass spectrometry in clinical practice: a comprehensive review of 16,175 samples. Mayo Clin Proc. 2020; 95(9):1852-64. Doi: 10.1016/j.mayocp.2020.06.029 PMid:32861330 [ Links ]
2. Gertz MA. Immunoglobulin light chain amyloidosis: 2022 update on diagnosis, prognosis, and treatment. Am J Hematol. 2022; 97(6): 818-29. Doi: 10.1002/ajh.26569 PMid:35429180 [ Links ]
3. Merlini G, Bellotti V. Molecular Mechanisms of Amyloidosis. New Engl J Med. 2003; 349(6): 583-96. Doi: 10.1056/NEJMra023144 PMid:12904524 [ Links ]
4. Merlini G. AL amyloidosis: from molecular mechanisms to targeted therapies. Hematology. 2017; 2017(1): 1-12. Doi: 10.1182/asheducation-2017.1.1 [ Links ]
5. Guan J, Mishra S, Qiu Y, Shi J, Trudeau K, Las G, et al. Lysosomal dysfunction and impaired autophagy underlie the pathogenesis of amyloidogenic light chain-mediated cardiotoxicity. EMBO Mol Med. 2014; 6(11): 1493-507. Doi: 10.15252/emmm.201404190 [ Links ]
6. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016; 8(6): 595-608. Doi: 10.15252/emmm.201606210 [ Links ]
7. Vaxman I, Dispenzieri A, Muchtar E, Gertz M. New developments in diagnosis, risk assessment and management in systemic amyloidosis. Blood Rev. 2020; 40: 100636. Doi: 10.1016/j.blre.2019.100636 [ Links ]
8. Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv. 2018; 2(10): 1046-53. Doi: 10.1182/bloodadvances.2018016402 [ Links ]
9. Kyle RA, Larson DR, Kurtin PJ, Kumar S, Cerhan JR, Therneau TM, et al. Incidence of AL Amyloidosis in Olmsted County, Minnesota, 1990 through 2015. Mayo Clin Proc. 2019; 94(3): 465-71. Doi: 10.1016/j.mayocp.2018.08.041 [ Links ]
10. Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O´Fallon WM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992; 79(7): 1817-22. Doi: 10.1182/blood.V79.7.1817.bloodjournal7971817 [ Links ]
11. Wisniowski B, McLeod DSA, Adams R, Harvey Y, Brown I, McGuire L, et al. The epidemiology of amyloidosis in Queensland, Australia. Br J Haematol. 2019; 186(6): 829-36. Doi: 10.1111/bjh.16000 PMid:31148162 [ Links ]
12. Pinney JH, Smith CJ, Taube JB, Lachmann HJ, Venner CP, Gibbs SDJ, et al. Systemic amyloidosis in England: an epidemiological study. Br J Haematol. 2013; 161(4): 525-32. Doi: 10.1111/bjh.12286 [ Links ]
13. Duhamel S, Mohty D, Magne J, Lavergne D, Bordessoule D, Aboyans V, et al. Incidence and prevalence of light chain amyloidosis: a population-based study. Blood. 2017; 130(Supplement 1): 5577-5577. Doi: 10.1182/blood.V130.Suppl_1.5577.5577 [ Links ]
14. Kumar N, Zhang NJ, Cherepanov D, Romanus D, Hughes M, Faller D V. Global epidemiology of amyloid light-chain amyloidosis. Orphanet J Rare Dis. 2022; 17(1): 278. Doi: 10.1186/s13023-022-02414-6 [ Links ]
15. Hasib Sidiqi M, Gertz MA. Immunoglobulin light chain amyloidosis diagnosis and treatment algorithm 2021. Blood Cancer J. 2021; 11(5): 90. Doi: 10.1038/s41408-021-00483-7 [ Links ]
16. Palladini G, Milani P, Merlini G. Novel strategies for the diagnosis and treatment of cardiac amyloidosis. Expert Rev Cardiovasc Ther. 2015; 13(11): 1195-211. Doi: 10.1586/14779072.2015.1093936 [ Links ]
17. Palladini G, Schönland S, Merlini G, Milani P, Jaccard A, Bridoux F, et al. The management of light chain (AL) amyloidosis in Europe: clinical characteristics, treatment patterns, and efficacy outcomes between 2004 and 2018. Blood Cancer J. 2023; 13(1): 19. Doi: 10.1038/s41408-023-00789-8 [ Links ]
18. Palladini G, Sachchithanantham S, Milani P, Gillmore J, Foli A, Lachmann H, et al. A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood. 2015; 126(5): 612-5. Doi: 10.1182/blood-2015-01-620302 [ Links ]
19. Kastritis E, Leleu X, Arnulf B, Zamagni E, Cibeira MT, Kwok F, et al. Bortezomib, Melphalan, and Dexamethasone for Light-Chain Amyloidosis. J Clin Oncol. 2020; 38(28): 3252-60. Doi: 10.1200/JCO.20.01285 [ Links ]
20. Gertz MA, Lacy MQ, Dispenzieri A, Buadi FK, Dingli D, Hayman SR, et al. Stem cell transplantation compared with melphalan plus dexamethasone in the treatment of immunoglobulin light-chain amyloidosis. Cancer. 2016; 122(14): 2197-205. Doi: 10.1002/cncr.30051 [ Links ]
21. Kastritis E, Palladini G, Minnema MC, Wechalekar AD, Jaccard A, Lee HC, et al. Daratumumab-Based Treatment for Immunoglobulin Light-Chain Amyloidosis. N Engl J Med. 2021; 385(1): 46-58. Doi: 10.1056/NEJMoa2028631 [ Links ]
22. Sidana S, Tandon N, Gertz MA, Dispenzieri A, Ramirez-Alvarado M, Murray DL, et al. Clinical features, laboratory characteristics and outcomes of patients with renal versus cardiac light chain amyloidosis. Br J Haematol. 2019; 185(4): 701-7. Doi: 10.1111/bjh.15832 [ Links ]
23. Muchtar E, Dispenzieri A, Wisniowski B, Palladini G, Milani P, Merlini G, et al. Graded Cardiac Response Criteria for Patients With Systemic Light Chain Amyloidosis. J Clin Oncol. 2023; 41(7): 1393-403. Doi: 10.1200/JCO.22.00643 [ Links ]
24. Kourelis T V, Kumar SK, Gertz MA, Lacy MQ, Buadi FK, Hayman SR, et al. Coexistent multiple myeloma or increased bone marrow plasma cells define equally high-risk populations in patients with immunoglobulin light chain amyloidosis. J Clin Oncol. 2013; 31(34): 4319-24. Doi: 10.1200/JCO.2013.50.8499 [ Links ]
25. Vaxman I, Sidiqi MH, Al Saleh AS, Kumar S, Muchtar E, Dispenzieri A, et al. Depth of response prior to autologous stem cell transplantation predicts survival in light chain amyloidosis. Bone Marrow Transplant. 2021; 56(4): 928-35. Doi: 10.1038/s41409-020-01136-2 [ Links ]
26. Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): A consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis. Am J Hematol. 2005; 79(4): 319-28. Doi: 10.1002/ajh.20381 [ Links ]
27. Comenzo RL, Reece D, Palladini G, Seldin D, Sanchorawala V, Landau H, et al. Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia. 2012; 26(11): 2317-25. Doi: 10.1038/leu.2012.100 [ Links ]
28. Posadas-Martinez ML, Aguirre MA, Brulc E, Saez MS, Sorroche P, Machnicki G, et al. Treatment patterns and outcomes in light chain amyloidosis: An institutional registry of amyloidosis report in Argentina. PLoS One. 2022; 17(10): e0274578. Doi: 10.1371/journal.pone.0274578 [ Links ]
29. Peña C, González JT, López-Vidal H, Donoso J, Contreras C, Vergara CG, et al. AL amyloidosis in the Chilean public health system: a pending debt. Multicenter study of the Chilean Monoclonal Gammopathies Cooperative Group. Rev Med Chil. 2019; 147(10): 1239-46. Doi: 10.4067/s0034-98872019001001239 [ Links ]
30. Michael M, Kastritis E, Delimpassi S, Michalis E, Repoussis P, Kyrtsonis MC, et al. Clinical Characteristics and Outcome of Primary Systemic Light-Chain Amyloidosis in Greece. Clin Lymphoma Myeloma Leuk. 2010; 10(1): 56-61. Doi: 10.3816/CLML.2010.n.006 [ Links ]
31. He D, Guan F, Hu M, Zheng G, He J, Han X, et al. The clinical characteristics and prognosis of chinese patients with light-chain amyloidosis: a retrospective multicenter analysis. Indian J Hematol Blood Transfusion. 2022; 38(3): 444-53. Doi: 10.1007/s12288-021-01469-y [ Links ]
32. Muchtar E, Gertz MA, Kyle RA, Lacy MQ, Dingli D, Leung N, et al. A Modern Primer on Light Chain Amyloidosis in 592 Patients With Mass Spectrometry-Verified Typing. Mayo Clin Proc. 2019; 94(3): 472-483. Doi: 10.1016/j.mayocp.2018.08.006 [ Links ]
33. Zhao Q, Li F, Song P, Zhou X, Wang L, Yu Y, et al. Clinical Characteristics and Treatment Outcome of Chinese Patients With Systemic Amyloid Light-Chain Amyloidosis: A Retrospective Single-Center Analysis. Clin Lymphoma Myeloma Leuk. 2016; 16(2): 104-10. Doi: 10.1016/j.clml.2015.11.001 [ Links ]
34. Lilleness B, Ruberg FL, Mussinelli R, Doros G, Sanchorawala V. Development and validation of a survival staging system incorporating BNP in patients with light chain amyloidosis. Blood. 2019; 133(3): 215-23. Doi: 10.1182/blood-2018-06-858951 [ Links ]
35. Palladini G, Milani P, Merlini G. Predicting survival in light chain amyloidosis. Haematologica. 2019; 104(7): 1294-6. Doi: 10.3324/haematol.2019.218859 [ Links ]
36. Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012; 30(9): 989-95. Doi: 10.1200/JCO.2011.38.5724 [ Links ]
37. Gillmore JD, Wechalekar A, Bird J, Cavenagh J, Hawkins S, Kazmi M, et al. Guidelines on the diagnosis and investigation of AL amyloidosis. Br J Haematol. 2015; 168(2): 207-18. Doi: 10.1111/bjh.13156 [ Links ]
38. Foli A, Palladini G, Caporali R, Verga L, Morbini P, Obici L, et al. The role of minor salivary gland biopsy in the diagnosis of systemic amyloidosis: results of a prospective study in 62 patients. Amyloid. 2011; 18(sup1): 80-2. Doi: 10.3109/13506129.2011.574354029 [ Links ]
39. Basset M, Milani P, Nuvolone M, Benigna F, Rodigari L, Foli A, et al. Sequential response-driven bortezomib-based therapy followed by autologous stem cell transplant in AL amyloidosis. Blood Adv. 2020; 4(17): 4175-9. Doi: 10.1182/bloodadvances.2020002219 [ Links ]
Recibido: 04 de Julio de 2023; Revisado: 30 de Julio de 2023; Aprobado: 24 de Septiembre de 2023
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
