











 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Remark
| 1) Why was this study conducted? |
| To present a series of cases from a National Reference Hospital using the current Diagnostic criteria for this disease, given that updated data on this pathology is poorly reported. |
| 2) What were the most relevant results of the study? |
| - Of the cases presented, 80% were men; - The average age of presentation is 65 years; - The duration of time from diagnosis to death was 6.5 months; - The early clinical manifestations in our study were cognitive and behavioral; - Our case series 100% of them presented a FIRDA pattern (intermittent frontal delta activity) followed by biphasic sharp waves 33%, triphasic sharp waves 50%. |
| 3) What do these results contribute? |
| Expansion of knowledge regarding this pathology and its clinical presentation forms as well as findings in auxiliary tests. |
Introduction
Creutzfeldt-Jakob disease (CJD) is a progressive and fatal neurodegenerative disease caused by the conversion of the normal brain prion protein (the cellular form of the prion-related protein PrPC) into a misfolded form, the scrapie protein (PrPSc) 1.
There are three main groups: sporadic CJD, genetic CJD and acquired CJD. Sporadic CJD (sCJD) is the most common and accounts for about 85% of cases. Genetic forms account for 10-15 %, including familial CJD, fatal familial insomnia and Gerstmann-Schäussler-Scheinker syndrome. Acquired forms account for 1-5 % including Kuru (related to historical ritual cannibalism in Papua New Guinea), iatrogenic CJD (iCJD) and variant CJD (vCJD) 2.
The overall incidence of CJD is estimated to be 1 to 2 cases per million per year and during the last 20 years this incidence may have increased, which could be explained by improved diagnostic methods. Most of them occur in late adulthood, between 50 and 70 years of age 3.
Variable clinical presentations can make it difficult to make a confident diagnosis. Necropsy remains the gold standard for definitive diagnosis. Diagnosis is based on a careful clinical history, physical examination to look for classic symptoms and signs, exclude other possible causes, and supportive diagnostic tests including brain magnetic resonance imaging (MRI), electroencephalogram (EEG), cerebrospinal fluid (CSF) 14-3-3 protein study and genetic testing 4.
There is no official registry in Peru that counts these patients, so each institution provides reports from time to time according to the frequency of this pathology. The main objective of the present study is to identify the main clinical manifestations and diagnostic methods of a series of cases of patients who met the criteria for probable sCJD, attended in a national reference hospital.
Presentation of cases
Case 1
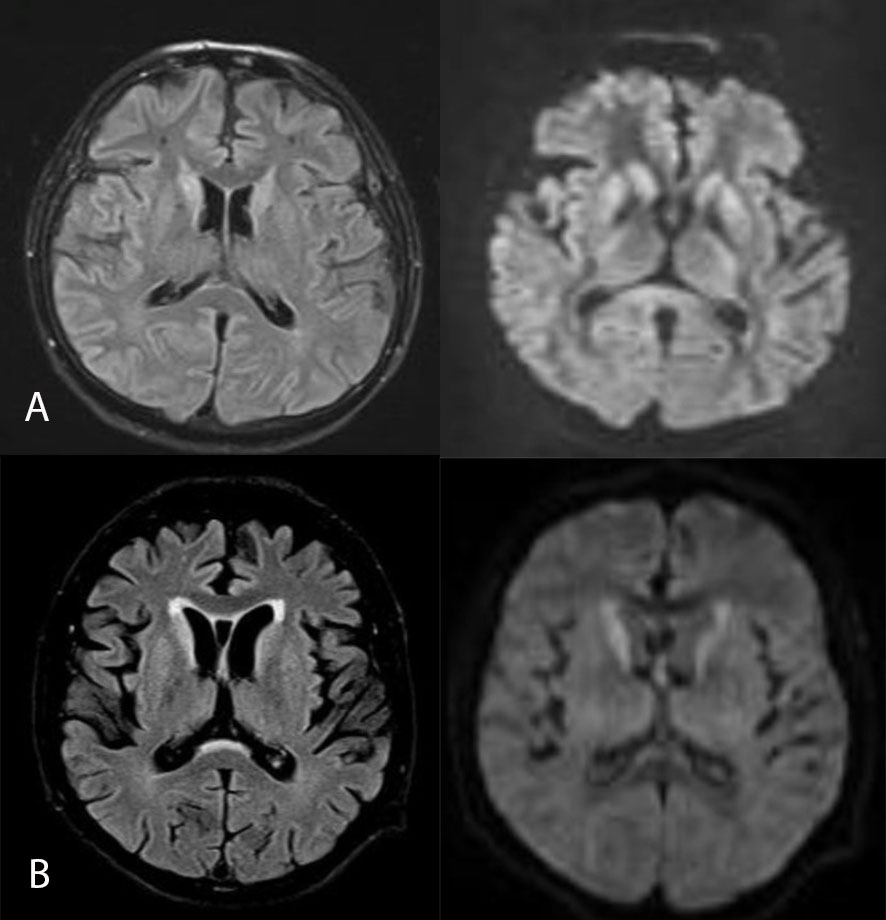
63-year-old male, with a medical history of hypothyroidism, hepatitis B and intestinal amebiasis, with an illness duration of one month, characterized by progressive instability for walking and grasping objects, in addition to visual hallucinations and episodes of fluctuating confusion. On physical examination, the patient was awake, inattentive, oriented only in person, partially obeying orders, left hemiparesis, global hypokinesia, preserved osteotendinous reflexes, positive prehension reflex, postural and kinetic tremor predominantly in the left upper limb, no dysmetria, no meningeal signs. Three weeks later, myoclonus in upper limbs predominantly right, laryngeal stridor, gaze with tonic deviation to the left, rapidly progressive deterioration of the content of consciousness, spasticity and bilateral hyperreflexia, akinetic mutism. In view of the clinical picture, she completed studies with brain MRI which showed hyperintensity of the head of the caudate nucleus and putamen, with slight restriction in diffusion, in addition to atrophy of frontotemporal predominance (Figure 1), as well as EEG and positive result for protein 14.3.3 in cerebrospinal fluid. She died 5 months after respiratory complications.
Case 2
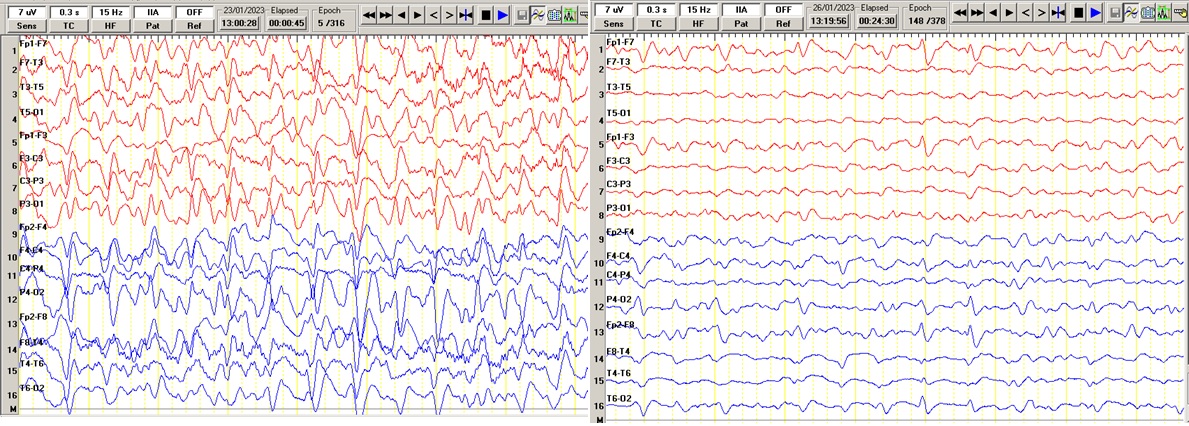
62-year-old woman, without pathologic history, with illness time of one month and a half, characterized by short-term alteration of the content of consciousness, attention deficit, dysphoria and emotional lability. In addition, progressive gait instability and negativistic childish behavior. On physical examination, the patient was awake, inattentive, decreased concentration, oriented only in person, hypofluent language, partially obeyed simple commands, mobilized the limbs, presented hypertonia, hyperreflexia and multisegmental myoclonias in four limbs, predominantly in the left hemibody. Two weeks later the patient evolved to total functional dependence, requiring a nasogastric tube for feeding. Subsequently, akinetic mutism was added, with accentuation of hypertonia and myoclonias. She had EEG (Figure 2A) and CSF 14.3.3 protein dosage. She died 3 months after respiratory complications.
Case 3
79-year-old male, with a history of arterial hypertension, with one month of illness characterized by progressive intermittent disorientation and alteration of recent memory, in addition to progressive gait instability and vertigo. On physical examination, the patient was awake, inattentive, oriented, concentration decreased, language preserved, muscle strength preserved, with evidence of multisegmental myoclonus in 4 extremities, without pyramidal signs. Two weeks later hypofluent language, hyponymia, mild akinesia and rapidly progressive deterioration of the content of consciousness adding multisegmental myoclonias. Subsequently progresses to superficial coma, increased myoclonus in lower limbs, as well as fasciculations in 4 limbs. She had supporting examinations including EEG (Figure 2B) and died after 6 months of illness due to respiratory failure secondary to pneumonia.
Case 4
Male 71 years old, without pathological history, with 2 months of illness, characterized by progressive gait instability and weight loss of 10 kg, plus dysarthria, dressing apraxia and recent memory disorder. On examination, the patient was awake, inattentive, not concentrated, with null abstraction and frontal release reflexes present, hypofluent language, preserved muscle strength, with dysmetria in extremities, bilateral extensor plantar reflex and clonus. In addition, there was evidence of fasciculations in the lower limbs, predominantly in the thighs. Studies were completed with MRI of the brain showing cortical atrophy predominantly mesial temporal and bilateral frontal, periventricular hyperintensity in frontal poles, lacunar hyperintensities of the radiate coronas of micro ischemic aspect with diffusion restriction (Figure 1), EEG and CSF, protein 14.3.3. Clinical picture progresses, dying 1 year after the beginning of the clinical picture due to respiratory complications and prostration state.
Case 5
69-year-old male, with no medical history, with one month of illness, characterized by short-term memory impairment, headaches, apathy and tendency to drowsiness. Subsequently, disorientation, balance disturbance, hypofluent language and gait instability were added, as well as rapidly progressive deterioration of the content of consciousness. On clinical examination, the patient was awake, inattentive, with akinetic mutism, with reactivity to deep nociceptive stimulus (pain gestures and withdrawal to the stimulus), preserved muscle strength, persistent cephalic and left brachial myoclonus, presence of bilateral extensor plantar reflex, no meningeal signs. Patient during hospital stay presents with respiratory complications, died 1 year after the onset of clinical picture.
Case 6
A 58-year-old woman with a medical history of arterial hypertension and diabetes mellitus, with a 7-month illness characterized by short-term memory impairment, irritability, emotional lability and insomnia. Subsequently, she had episodes of unmotivated laughter and difficulty swallowing. On physical examination she is awake, hypoattentive, does not obey simple commands, episodes of unmotivated laughter, anosognosia, preserved muscle strength, hyperreflexia, extensor plantar cutaneous reflex, cautious gait. After 3 weeks the picture progresses to akinetic mutism. She died 2 months later.
Discussion
CJD is a progressive and fatal neurodegenerative disease that is considered rare due to its incidence 5,6. We present six patients (Table 1), predominantly male, with a mean age of 70 years. The main clinical manifestation was progressive cognitive impairment, within the approach to a rapidly progressive dementia it is necessary to rule out other pathologies, in our institution metabolic, infectious, tumor, autoimmune and drug diseases were ruled out. During the course of their studies, other symptomatologies were added, such as myoclonias, motor and cerebellar symptoms, and almost all of them ended up with mutism 7,8.
Table 1 Clinical and laboratory characteristics of patients.
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | |
|---|---|---|---|---|---|---|
| Age (years) | 63 | 62 | 79 | 71 | 69 | 58 |
| Sex | M | F | M | M | M | F |
| Time from illness to death (months) | 5 | 3 | 6 | 6 | 12 | 9 |
| Signs and symptoms | Ataxia, cognitive impairment, visual hallucinations, myoclonias | Ataxia, cognitive impairment akinetic mutism, myoclonias | Cognitive impairment, Confusion, ataxia, myoclonias | Ataxia, confusion, cognitive impairment, fasciculations | Cognitive impairment, myoclonias | Cognitive impairment, confusion, akinetic mutism |
| EEG | Generalized slowed baseline activity, without evidence of epileptiform discharges and periodic activity. | Slowed baseline activity in the theta range, medium to high voltage irregular, with interposition of diffuse and accentuated three-phase periodic activity in anterior right segments | Slowed baseline activity, bilateral periodic activity, with acute triphasic waves of medium to high voltage at 0.8 -1.2 Hz, right parasagittal predominance | Generalized slowed baseline activity, without evidence of epileptiform discharges and periodic activity. | Slowed base activity with three-phase waves at 0.5-1.5 Hz on severely slowing background. | Generalized and disorganized baseline slowed activity without epileptiform activity or periodic activity. |
| LCR (Protein 14 3.3) | Positive | Positive | Not performed | Positive | Not performed | Positive |
| Cytochemical, and unaltered cultures | Cytochemical, and unaltered cultures | Cytochemical, and unaltered cultures | Cytochemical, and unaltered cultures | Cytochemical, and unaltered cultures | Cytochemical, and unaltered cultures |
M: Male, F: Female, EEG: Electroencephalogram, RM: Magnetic Resonance, LCR: Cerebrospinal fluid
Conclusion
In our environment, sCJD has a varied clinical presentation, manifesting most frequently with rapidly progressive dementia, followed by myoclonias with subsequent evolution to akinetic mutism. This type of patient is a diagnostic challenge, which implies clinical suspicion and exclusion of other etiologies. EEG, 14-3-3 protein in CSF and brain MRI contribute to the diagnosis. Currently there is no treatment for this entity and there is a high probability of death before one year.

