










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkBiosalud
Print version ISSN 1657-9550
Biosalud vol.9 no.1 Manizales Jan./June 2010
MECANISMOS INTRACELULARES INVOLUCRADOS EN EL APRENDIZAJE Y LA MEMORIA DEL MIEDO
INTRACELLULAR MECHANISMS INVOLVED IN THE LEARNING AND MEMORY OF FEAR
Juan David Sánchez-Ramírez1 y Luis Fernando Uribe-Velásquez2
1 Estudiante MVZ. Universidad de Caldas. Facultad de Ciencias Agropecuarias. Programa de Medicina Veterinaria y Zootecnia. Manizales, Colombia. Correspondencia: judasra@gmail.com.
2 Ph.D. Universidad de Caldas. Facultad de Ciencias Agropecuarias. Departamento de Salud Animal. Manizales, Colombia.
Recibido: septiembre 25 del 2009 - Aceptado: enero 19 del 2010
RESUMEN
La neurobiología del miedo consiste en una amplia configuración celular que implica la actividad en conjunto de un gran número de neuronas. Tales conexiones sufren cambios a lo largo de la vida en un proceso dependiente de la actividad celular. Esto hace variar la efectividad de la comunicación sináptica, facilitando el desencadenamiento del miedo. Por eso, esta revisión tiene como fin describir los procesos fisiológicos causantes de ese cambio celular en el miedo, los cuales inician con la activación de receptores iónicos y metabotrópicos, para finalizar con la estimulación genómica y la síntesis de proteínas. Así mismo, exponer la relación del miedo mientras se establecen memorias asociadas con él, como un factor que contribuye a una alta frecuencia de descarga y despolarización celular que favorece los cambios a largo plazo debido a la intensa excitación neuronal. Se concluye que, neurobiológicamente, el miedo puede fortalecerse luego de estimulaciones aversivas, mediante la formación de asociaciones entre estímulos y, a su vez, de éstos con el contexto, lo que le propicia al organismo una reactividad más eficiente frente a un encuentro posterior con la misma amenaza o circunstancia.
PALABRAS CLAVE: aprendizaje, memoria, miedo, sinapsis, proteínas quinasas, segundos mensajeros, factores de transcripción, genes.
ABSTRACT
The neurobiology of fear is a large cellular configuration that implies the group activity of a large number of neurons. These connections suffer changes along the life cycle in a cellular activity-dependant process. This changes the effectiveness of the synaptic communication, facilitating the unchaining process of fear. Hence, the objective of this review is to describe the physiological process that cause those changes in fear, which begin with the activation of ionics and metabotropics receptors, and ending with the genomic stimulation and protein synthesis. Additionally, this paper explains the relation of fear while memories associated with it are established, as a factor that contributes to a higher frequency of discharging and cellular depolarization that favors long term changes due to intense neural excitation. In conclusion, fear can be neurobiologically strengthened after aversive stimulations, by the formation of associations between stimuli, as well as between them and the context, encouraging the organism to have a more efficient reactivity regarding a later meeting with the same threat or circumstance.
KEY WORDS: learning, memory, fear, synapse, protein kinases, second messengers, transcription factors, genes.
EL MIEDO EN EL APRENDIZAJE Y LA MEMORIA
Neurobiológicamente, el miedo es definido como el entramado de conexiones neuronales establecidas en circunstancias de peligro o amenaza (1, 2). Esa capacidad reactiva propicia un ahorro de tiempo en momentos que requieren una rápida respuesta, pudiendo ser determinante para un desenlace a favor o en contra de la supervivencia de cada especie. Por ello, esta emoción está encaminada a proteger el organismo de posibles daños (1, 2). Sin embargo, sumado a esto, es importante ir haciendo más eficientes las reacciones emocionales en situaciones posteriores, en las que se presenta el mismo estímulo o un contexto similar; pues de no ser así, se correrían repetidamente los mismos riesgos, al no sacar provecho de la experiencia previa. Es aquí donde además de una capacidad para actuar ante circunstancias de peligro, se puede llegar también a relacionar las amenazas con las circunstancias concretas, en las cuales se desenvuelve un organismo, y de esta manera, actuar prontamente en un encuentro posterior.
El miedo puede entonces ser aprendido, y da una consecuente respuesta más rápida y selectiva ante la posibilidad de un nuevo encuentro con la misma amenaza. Este aprendizaje y memoria emocional permite a lo largo de la vida enriquecer las relaciones que el animal establece entre estímulos y respuestas. La memoria del miedo ha sido clasificada como un tipo de memoria implícita o no declarativa (3-5) que se caracteriza por ser inconsciente, es decir, la asociación entre estímulos ocurre sin el animal darse cuenta. El condicionamiento clásico (3, 5, 6) es un proceso de relación que explica cómo ocurre este tipo de memoria. Éste conlleva a desencadenar conductas luego de aparear de manera consecutiva un estímulo que cause una respuesta con otro que no; la asociación entre ambos estímulos se establece debido a la cercanía temporal y la periodicidad con la cual se presentan (5, 7-12). Tal condicionamiento se acomoda al modo como ocurre el aprendizaje en el miedo, ya que puede ser suscitado bajo la presentación de un estímulo innato que signifique peligro para el animal, como un pinchazo eléctrico sobre la piel o la presencia de un predador natural, en conjunto con otro estímulo que no signifique algo para el animal, como un tono o una luz; con el tiempo, esa relación permite la anticipación del suceso aversivo o nocivo -estímulo innato-, luego de la presentación del estímulo que en un comienzo no significaba nada (10, 13-16).
En este proceso se ha observado actividad neuronal en el núcleo basolateral, lateral, medial y central de la amígdala (3, 5, 6). Los núcleos lateral y medial sirven como puntos de convergencia de ambos estímulos, y el núcleo central sirve como zona de proyección desde la amígdala hasta las áreas de control motor, autónomo y endocrino. Aquellos estímulos que signifiquen peligro estimularán el núcleo central, mientras que aquellos inocuos inhibirán esta actividad de proyección por la activación de masas de interneuronas inhibitorias (1, 2). Sin embargo, la cercanía temporal entre los dos estímulos comienza a reforzar la conectividad y funcionalidad sináptica del estímulo inocuo, que adquiere la capacidad, él solo, de desencadenar excitación en el núcleo central y las respuestas derivadas (17-20). Esto comienza a vislumbrar, además de lo comentado, que existe una propiedad morfofisiológica que podría ser la base orgánica para el aprendizaje y la memoria.
Por otro lado, el miedo también ha sido relacionado con otro tipo de memoria encargada de conservar los datos sobre los hechos, eventos o sucesos, denominada memoria explícita o declarativa (3-5). En este caso, igualmente se presenta una asociación, pero en vez de ser entre estímulos por separado se tiene en cuenta el contexto donde se desenvuelve el organismo, o sea, aquellos estímulos simultáneos que ayudan a dimensionar el evento. Esto es posible al aparear el estímulo incondicionado con un contexto, y no con otro estímulo en particular. De manera que la situación no placentera será construida a partir del conjunto de estímulos percibidos en el lugar y su relación con la amenaza. La característica de estas asociaciones es que son impredecibles, pues no se relaciona el estímulo nocivo con otro en particular, sino con varios a la vez, lo cual hace indeterminable un orden de ocurrencia entre ellos y el estímulo adverso (5, 12).
En este caso, tal tipo de condicionamiento implica la participación de la amígdala en conjunto con el hipocampo. Ambas estructuras se han identificado como responsables de la asociación del estímulo incondicionado con el contexto de importancia para identificar la situación donde se presentará el estímulo nocivo (1, 5, 21). También se han observado cambios funcionales en la corteza prefrontal, en donde disminuye la actividad neuronal, potenciando la actividad en la amígdala al dejar de ser estimuladas las interneuronas inhibitorias presentes en esta última (22). De esta forma, las neuronas del núcleo lateral, basal y basolateral de la amígdala, las células de la región CA1, CA3 y el giro dentado del hipocampo, poseen propiedades funcionales que brindan la posibilidad de reforzar sus conexiones a lo largo de la vida (1, 3, 9, 17, 18, 20, 23). Un tercer tipo de condicionamiento será mencionado más adelante, y se caracteriza por realizar asociaciones más débiles y quebrantables en el tiempo.
Cada estructura neuroanatómica aporta una parte de la red neuronal que conforma el miedo, y está constituida por un gran número de células (1, 2). La conectividad entre múltiples neuronas puede modificarse mediante las asociaciones mencionadas anteriormente (los condicionamientos). Esto conlleva a que se presenten mensajes químicos entre las células nerviosas, a la vez que ocurren también en su interior, siendo los responsables del cambio funcional. Es la actividad, en diferentes momentos y lugares, la que refuerza unas conexiones y no otras. Por ello, dependiendo de esta acción, se irán modificando las reacciones celulares.
En cada célula puede haber cambios fisiológicos a corto plazo (minutos a horas), no requiriendo de síntesis proteica o ARNm. Esa variación es una modificación temporal de moléculas existentes que pasan de un estado inactivo a uno activo. Entre éstas se encuentran enlaces covalentes o translocaciones proteicas. A su vez, los cambios a largo plazo (horas a días) sí precisan de nueva síntesis ARNm y proteica. De esta manera, la historia del individuo construye biológicamente la memoria del miedo a través de varios mecanismos celulares y moleculares (7, 9, 11, 24-26).
La nueva capacidad de respuesta celular influye en la comunicación intercelular. Una neurona activa aumenta la probabilidad de desencadenar la acción de otras con las que tiene sinapsis. En las sinapsis químicas, caracterizadas por poseer moléculas transmisoras que se liberan al espacio extracelular, las moléculas transmisoras son entonces las gestoras de las modificaciones intracelulares, tanto en la célula presináptica como en la postsináptica (18, 27-31), las cuales facilitarán la transmisión del impulso nervioso de neurona a neurona. Los cambios que conllevan a una mayor eficiencia en la comunicación neuronal ocurren gracias a la actividad coherente entre dos neuronas conectadas. En otras palabras, el cambio sucede cuando la célula presináptica está o ha liberado neurotransmisores a la hendidura sináptica, y cuando la célula postsináptica, en ese mismo momento, posee cierto grado de despolarización. La apertura de más canales iónicos intensifica la inversión en la carga intracelular, desencadenando un conjunto de reacciones en cadena que dan marcha al reforzamiento.
Mediante procedimientos in vitro es posible conseguir esa eficiencia, que se conoce como potenciación a largo plazo (PLP). Ésta se consigue cuando una neurona previamente despolarizada recibe varias veces una estimulación consecutiva. Algunos autores han sugerido que este fenómeno refleja celularmente cómo puede llevarse a cabo el aprendizaje (32). No obstante, otros autores reportan que PLP y el condicionamiento del miedo no son procesos homólogos, a pesar de que implican un sustrato bioquímico igual y las mismas células en el núcleo lateral de la amígdala. Mencionan, además, que una diferencia consiste en el decaimiento a largo plazo de la eficacia sináptica, siendo más rápidos en la PLP que en la memoria del condicionamiento (17, 20). Es probable que la diferencia anotada anteriormente consista, por una parte, en el número de neuronas implicadas en un proceso u otro, pues en la PLP participa un pequeño número de neuronas, mientras que en el condicionamiento actúan poblaciones. Además, se puede presentar una interconexión más compleja en el condicionamiento, lo que hace más probable la excitación y la modulación de una neurona en particular al ser más amplio el número de entradas sinápticas que en ella convergen. A pesar de esto, la PLP ha sido empleada para la identificación de las moléculas y sus funciones ocurridas en el aprendizaje del miedo.
Para que la memoria alcance una duración a largo plazo, los mecanismos requeridos consisten en el curso de procesos con características diferentes. Como parte de ellos, está la consolidación de la memoria, que se refiere a la adquisición de la misma. La reconsolidación, como otro proceso, aún se debate entre tres tópicos: la recapitulación de la memoria adquirida, el componente tardío de esa consolidación o la recuperación posterior de la memoria para su fijación por largos periodos. Estos procesos poseen mecanismos comunes, tales como cambios temporales de la actividad molecular o la síntesis ARNm y proteína (33, 34), y aunque algunos reporten que la reconsolidación no requiere de tal síntesis, sin importar la intensidad del entrenamiento o su antigüedad (33), la mayoría de estudios relatan este suceso común (3, 9, 18, 21, 23, 30, 31, 35). Sin embargo, no todas las estructuras anatómicas envueltas en la consolidación se encuentran envueltas en la reconsolidación (33, 34), fundamentando anatómicamente diferencias entre cada mecanismo. Otra diferencia que salta a la vista es el tiempo en que se presentan los cambios funcionales y su duración. En una escala menor, la consolidación y reconsolidación son interacciones moleculares organizadas en complejas cadenas de reacciones. Es más, no es claro el papel que algunas moléculas muestran en algunos de estos eventos como la adquisición. Tal es el caso, por ejemplo, de los receptores tipo NMDA y no NMDA, que al bloquearse de forma selectiva cualquiera de ellos, impide la adquisición del aprendizaje en entrenamientos cortos. Sin embargo, cuando se realizan entrenamientos largos este suceso se revierte; en cambio, cuando se bloquean los dos tipos de receptores a la vez, la alteración se manifiesta hasta en los entrenamientos largos (36). De esta forma, se evidencia el gran número de factores que pueden influir en un aprendizaje del miedo y los cambios en el proceso fisiológico cuando alguno de ellos se modifica.
La actividad de cada sustancia depende de la variación producida en su estructura al relacionarse con otras moléculas. Todo inicia con el movimiento iónico, causante de la despolarización. La intensidad de esa despolarización estará directamente relacionada con la magnitud de los cambios. Luego, la actividad neuronal da paso a una fase temprana (1 a 3 horas), en la que ocurren modificaciones covalentes de proteínas existentes, sin presentar síntesis proteica. Posteriormente, sobreviene una fase tardía (horas a días) que implica síntesis de nuevas proteínas mediante segundos mensajeros (como el AMPc) y la acción de quinasas como proteína quinasa A (PKA), proteína quinasa mitógeno activa (MAPK), entre otras (9, 23, 37).
Se ha observado que la intensidad del entrenamiento, en el condicionamiento contextual, influye la fase tardía de la potenciación a largo plazo (24, 37). Un fuerte entrenamiento presenta un solo evento de síntesis proteica, causado por la activación de varias vías moleculares a la vez, producto de la alta frecuencia de despolarización, que inician una gran expresión génica que da lugar a una síntesis proteica masiva; inclusive, se ha reportado que un sobre-entrenamiento por varios días puede oponerse a un daño en la amígdala antes del condicionamiento pavloviano, y permitir la adquisición del aprendizaje relacionado con el miedo (36). Ese suceso molecular múltiple permite una rápida consolidación. Por su parte, en el entrenamiento débil ocurren dos eventos de síntesis antes de la PLP, ya que la expresión génica iniciada con el primer evento no es suficiente (37).
Otro hallazgo es que entre mayor sea el número de acciones neuronales que convergen en alguna zona particular, más difícil se hace consolidar la memoria. Por ejemplo, el condicionamiento clásico es fácilmente consolidado, en comparación con los condicionamientos de segundo orden, en los que el primer estímulo condicionado se asocia con un segundo estímulo condicionado, y así sucesivamente. Las asociaciones neuronales más numerosas, correspondientes a relaciones de una mayor cantidad de estímulos, son más complejas pero más débiles y menos persistentes. Este último condicionamiento no posee cambios moleculares de larga duración, sino sólo translocaciones proteicas temporales, lo cual es inverso al condicionamiento clásico, que se fortalece por la síntesis proteica (38). El tipo de asociación y el grado de activación en el entrenamiento marcan la estabilización y durabilidad del aprendizaje emocional.
DESDE EL CITOPLASMA AL NÚCLEO, COMPONENTES MOLECULARES DE LA VÍA DEL AMPc
Los mecanismos moleculares descubiertos hasta hoy incluyen: receptores para las sustancias, proteínas quinasas A, C, MAPK, ERK, proteínas G, segundos mensajeros como el AMPc y el Ca2+, factores de trascripción tal como CREB (proteína elemento conjugadora de respuesta a AMPc), histonas y genes (28, 37, 39, 40). Para que se produzca actividad genética, se requiere que coactivadores multifuncionales y factores de transcripción actúen en varias reacciones de ocurrencia sincrónica (41) (Figura 1).
Una de las vías que se han identificado para la formación de memoria a corto y largo plazo es a través del AMPc. Ésta ocurre en la consolidación, y se observa en amplia variedad de especies. Produce, finalmente, síntesis proteica por medio de factores de transcripción que dan comienzo a la transcripción y posterior traducción (3, 9, 17, 27, 37, 42-45). Inicia con un receptor de membrana acoplado a una proteína G o directamente a la adenil ciclasa (como los receptores para dopamina D1/D5 o receptores β1 adrenérgicos). Después de la unión de un ligando extracelular al receptor acoplado a proteína G y por liberación de Ca+2 a través de la proteína Ca2+ calmodulina, G activa la enzima adenil ciclasa que sintetiza el AMPc a partir de AMP. El AMPc estimulará la proteína quinasa A (PKA). En este punto, se pueden producir cambios bioquímicos temporales por fosforilación de proteínas y canales iónicos, requeridos en la memoria a corto plazo; o cambios a largo plazo, por estimulación genética (35, 37, 44, 46), al activar otras quinasas como ERK o fosforilar directamente las histonas (47). Ambas alternativas dependen de la intensidad en la despolarización y el aumento de Ca2+ intracelular. Otra molécula activada por el AMPc es la proteína de intercambio activada por AMPc (Epac) o factor de intercambio de guanina/AMPc, relacionada con la recuperación de la memoria (reconsolidación) (35). Finalmente, el AMPc es degradado por una fosfodiesterasa de AMPc (46), por lo que frena la activación de PKA. Las interneuronas y astrocitos pueden modular la plasticidad y la memoria a través de esta vía del AMPc (44), aumentando o disminuyendo la actividad de una neurona y su respuesta a la estimulación.
La proteína G activadora de la adenil ciclasa posee una subunidad conocida como Gαs, que incrementa la actividad de esta molécula, y contrario a lo esperado, disminuye la actividad del AMPc. El aminoramiento de esa actividad se explica de manera directa e indirecta: 1) la activación del AMPc inicia la acción de PKA, lo que a su vez fosforila las fosfodiesterasas encargadas de la inactivación del segundo mensajero en una relación directamente proporcional, y 2) la activación genómica expresa dichas enzimas catalíticas (46). Por eso, al parar la acción del AMPc, Gαs causa déficit en la consolidación y recuperación de la memoria de tipo asociativa -condicionamiento-. En este caso, se identificó que la alteración en la memoria se correlacionaba con el exceso de actividad PKA inducido por la proteína G, porque esos efectos fueron revertidos luego de inhibir esta quinasa (46). Sin embargo, la actividad de PKA favorece la memoria al largo plazo cuando actúa por lapsos de tiempo. En contraparte, la supresión genética de moléculas como: carboxi terminal ubiquitina hidroxilasa, la cual funciona como unidad proteolítica de la subunidad reguladora de PKA, disminuye su regulación y produce una forma continuamente activa de PKA por 10 a 12 horas, tiempo necesario para que se lleven a cabo los cambios estructurales a largo plazo (37). En vista de esto, es posible que haya un límite temporal de actividad, en el cual la célula regule cada vía molecular para mantener un equilibrio entre activación e inactivación, y abrir periodos temporales en los que ocurren los cambios celulares.
El AMPc también activa al factor de transcripción proteína de respuesta de unión al AMPc (CREB) a través de PKA o MAPK. Esta proteína CREB, junto con otros factores de transcripción activadores e inhibidores, causa expresión de los genes CRE, codificando enzimas, receptores, proteínas citoesqueléticas, factores de transcripción y de crecimiento (9, 23, 42, 43, 45, 48). Dentro de los factores inhibidores, está la proteína de represión temprana de AMPc (ICER), que es un potente represor endógeno de los genes CRE por su afinidad a los dominios del ADN, causando su apagado. El balance entre CREB/ICER modula la síntesis proteica (43, 45). El aumento de ICER disminuye la potenciación a largo plazo, atenuando el aprendizaje y la memoria, con consecuencias negativas, especialmente frente aquellos estímulos débiles, y positivas, pues evita el exceso de plasticidad neuronal que conduciría a exageradas memorias de miedo o epileptogénesis (45). Esto indica que la inhibición acontece en varios momentos para regular los cambios a largo plazo, incluso, cuando tales reacciones en cadena han sido desatadas, estableciendo un rango funcional, en el cual un evento culmina o no con una nueva síntesis molecular. En contraste, un estudio reportó que la disminución de CREB no repercute sobre el condicionamiento contextual de miedo, pero sí sobre el condicionamiento aversivo a sabores (43). Ese hallazgo sugiere que en el condicionamiento del miedo pueden existir otras vías que agrupan factores diferentes a CREB.
Las vías alternas de CREB pueden darse por medio de otra proteína de unión a CREB (CBP); esta unión ocurre por medio del dominio KIX de ambas moléculas y favorece la acetilación de histonas con la consecuente expresión genética (41, 42). La proteína CBP es un coactivador de la proteína CREB involucrada en la formación de memoria a largo plazo (41, 42). CBP activa la maquinaria general para la transcripción genética al interactuar con múltiples reguladores transcripcionales y enzimas como la histona acetil transferasa (42, 48), y a su vez, también puede cumplir esa acción enzimática de acetilación por sí mismo (41, 42). Al final, igual a como lo hace CREB, el estado natural silente de la cromatina es removido, al alterarse su estructura reprimida epigenéticamente mediante la modificación de la carga de las histonas, colocándolas en estado activo (48). Por ello, la disminución de CBP altera el condicionamiento contextual dependiente del hipocampo (48).
LA FOSFORILACIÓN REALIZADA POR LAS KINASAS
Cuando ocurre una despolarización intensa, el Ca2+ ingresa a través de canales para calcio dependientes de voltaje y receptores N metil D aspartato (NMDA) (49-52), o es liberado de las zonas de almacenamiento intracelulares por la calmodulina, lo que hace que en conjunto con el AMPc, se produzca la activación de quinasas como PKA, MAPK y ERK (17, 53). La función de estas proteínas radica en fosforilar otras moléculas.
En invertebrados y vertebrados, se ha observado que PKA, MAPK y ERK se trasladan al núcleo para fosforilar proteínas mediadoras de transcripción como CREB e inducir expresión de los genes CRE, y de esta forma, transcribir de ADN a ARNm. Por ello, MAPK/ERK potencian la acción de PKA al fosforilar también el factor CREB (3, 9, 17, 37, 43, 44, 45, 47, 54, 55); además, ERK se comporta como factor de transcripción cuando se encuentra en el núcleo (21), y por ello contribuye de dos formas al fenómeno de síntesis proteica, como estimulador de CREB y como factor de transcripción. Tanto MAPK como ERK, a su vez, intervienen como señales extracelulares necesarias para que en el lado presináptico se desencadene el proceso transcripcional que permitirá la efectividad sináptica (3, 18). En el caso de MAPK, es una quinasa de serina-treonina extensamente expresada en dendritas y soma de neuronas. Regula la proliferación y diferenciación celular mediante la generación de segundos mensajeros (fosfolipasa A2), organización citoesquelética (fosforilación de proteína MAP2) y transcripción genética (56).
Algunos autores han observado que la inhibición de ERK no afecta ni la adquisición ni la memoria a corto plazo, y en cambio, sí la afecta a largo plazo, presentándose primero expresión en el córtex prefrontal y posteriormente -1 hora después- en el hipocampo (30). Por su parte, otros observaron que la inhibición de ERK en la corteza entorrinal provoca aumento de las conductas de congelamiento, lo que sugiere, según ellos, que estas proteínas participan de manera negativa en el condicionamiento contextual del miedo (57). Esto quiere decir que la acción de esta enzima depende del tipo de memoria y estructura evaluada, mostrando así que cada molécula puede tener una acción diferente, incluso contraria; además, no se puede descartar la posibilidad de que en algunas áreas predomine más la actividad bioquímica de otras quinasas o, en cambio, sea la forma en que se estimula la determinante de la actividad molecular intracelular.
Por otro lado, es de destacar que un estudio reporta una diferencia de género en el condicionamiento contextual de miedo, caracterizada por una mayor retención de la memoria a largo plazo en ratones machos en comparación con las hembras (58). La variación conductual observada fue correlacionada con una mayor expresión de ERK/MAPK en la región ventral del hipocampo de los machos. Esto refleja que las diferencias mnésicas de género radican en factores como la magnitud en la actividad de las quinasas en áreas determinadas.
Las Ca-Calmodulina protein kinasas II y IV (CaMK II o CaMK IV) igualmente participan en la memoria del condicionamiento del miedo y la plasticidad sináptica en el nivel postsináptico, en espinas dendríticas y los somas neuronales del núcleo lateral de la amígdala, sobre todo su porción dorsal donde llegan las proyecciones auditivas del tálamo, y en el hipocampo. Se ha hallado subtipos α, β, γ, δ. De este conjunto, el que más implicación tiene es el subtipo α. En cambio, el subtipo β ejerce un efecto contrario y los otros dos subtipos no se encuentran en el sistema nervioso. Durante la elevación intracelular de Ca2+, CaMKII se traslada a la sinapsis en donde puede interactuar y modular proteínas de densidad postsináptica, incluyendo los receptores AMPA y NMDA (11, 25, 29, 53, 59). No obstante, CaMKII también puede sufrir una autofosforilación en un residuo de treonina, transformándola en una forma persistentemente activa independiente de Ca2+ (29). Entre sus acciones, se destaca la apertura de los canales NMDA por fosforilación de su subunidad NR2B, por la que tiene alta afinidad (25, 29, 53). Otra es fosforilar la subunidad GluR1 del receptor AMPA, induciendo su inserción dentro de la sinapsis al permitir una interacción del dominio COOH de GluR1 con el dominio de alguna proteína PDZ (PSD 95, Dlg, ZO1); esta última ayuda a fijarlo a la superficie celular para cumplir con su actividad receptora (59). CaMKII también fosforila a CREB con la consecuente activación de genes CRE (11, 23) y a su coactivador CBP (48).
La proteína kinasa C (PKC) puede fosforilar histonas (48). Esta enzima tiene un dominio amino terminal con dos sitios para unión de segundos mensajeros: Ca2+ y diacilglicerol, así como una porción autoinhibitoria, además de un dominio carboxi terminal que contiene el sitio catalítico. En condiciones basales, la porción autoinhibitoria se liga a la porción catalítica manteniendo la enzima inactiva, cuando los segundos mensajeros se unen a uno de sus sitios, la liberan de su autoinhibición y la activan. Esta kinasa posee una isoforma: la proteín kinasa Mζ (PKMζ), la cual tiene la propiedad de permanecer activa sin una estimulación dada por segundos mensajeros, ya que le falta el dominio de autoinhibición (60). El papel que desempeña en el mantenimiento de la memoria está relacionado con los datos finos como la discriminación precisa entre memorias de localización espacial. La presencia de tales isoformas sin dominio de autoinhibición en el aprendizaje y memoria indica que son moléculas de alta importancia en el mantenimiento de los cambios funcionales, luego de la adquisición de la memoria, y por lo tanto, no se requieren para la adquisición de la misma, o sea, en el momento en que sucede el evento de aprender por primera vez.
EL FACTOR DE TRANSCRIPCIÓN NUCLEAR KAPPA
El factor nuclear kappa B (FNκB) hallado en células CA1 del hipocampo, actúa en la reconsolidación de la memoria. La activación del FNκB ocurre al desligar el dominio inhibitorio de κB (IκB) que mantiene bloqueada su capacidad de regulación en la transcripción; tal conjugación se rompe gracias a la fosforilación ejercida por la enzima IκB quinasa (IKK). Luego de esta liberación, el FNκB se dirige al núcleo y se liga a un elemento regulador de genes κB, que activan a la histona H3 e inician la transcripción. Una segunda vía es independiente del FNκB y se da por medio de subtipos de IKK y la activación de otras quinasas o acetilasas en vez de FNκB. La vía alterna se ejecuta por IKKα y finaliza con la acción del complejo histona acetiltransferasa y CBP, causando acetilación de H3 para promover la acción genética. En vista de eso, la inhibición de FNκB no reduce la actividad de H3 porque ésta es estimulada por la vía alterna de IKKα; en cambio, la inhibición de IKKα sí disminuye la acción de H3 (31). Es posible que, al ser el efecto de IKKα causado por el complejo acetilasa-CBP, su inhibición frene la acción del complejo, y como éste es punto de convergencia de otras vías (como AMPc y CREB), la alteración de su función tiene mayor repercusión y deprime entonces la activación final de la histona.
EL ESTADO ACTIVO E INHIBIDO DE LA CROMATINA
Todas las reacciones en cadena a través de quinasas y factores de transcripción mencionadas conducen a la modificación de histonas, reguladoras del cambio de estado inhibido de la cromatina -complejo proteico dinámico que empaqueta el ADN- a un estado transcripcional activo, que ayuda a regular la expresión genética presente en la memoria (48, 61, 62, 63). Las histonas son proteínas que hacen parte del bloque de construcción de la cromatina, que junto con una porción de ADN forman los nucleosomas -unidades de la cromatina- (42, 48); particularmente, la histona H3 es importante para el condicionamiento del miedo (48, 61-63). Con esos componentes se ha formulado una hipótesis, el código de histonas, que plantea un mecanismo epigenético para la expresión genética a partir de distintos patrones de modificación de una o más colas de las histonas, reclutando sitios específicos en esas moléculas que conducen a perfiles de expresión genética particulares (48, 63). Las colas de las histonas poseen dominios amino terminales que se extienden mas allá de su centro globular, las cuales orquestan la remodelación específica de la cromatina (48, 61). Las colas son fosforiladas por ERK, proteína quinasa 1 mitógena y estrés activa -adicionando grupos fosfatos negativos-, y acetiladas por la enzima histona acetil tranferasa, que ocasiona activación de las histonas (41, 42, 48, 61-63), al neutralizar la carga amino positiva de la cola, producto de su alto contenido de lisina (48, 63). Esto deja libre el ADN para la unión de enzimas (como RNA polimerasa II), factores de transcripción (CREB y Elk1 que guían al sitio de transcripción las RNA polimerasas), en apoyo de factores coactivadores (CBP) (63). Se ha visto que las histonas pueden conservar su modificación por algunos períodos después de ocurrida la transcripción, facilitando un evento transcripcional posterior (48). Luego, la acetilación y la fosforilación son revertidas por enzimas histonas deacetilasas (42, 48, 63) y fosfatasas, respectivamente, devolviendo la cromatina a su estado inicial de apagado (Figura 2).
El cambio, finalmente, influirá en el comportamiento neuronal y en las respuestas individuales de cada célula dentro de la red nerviosa a la que pertenece. Todas esas interacciones moleculares asegurarán que todo ese conjunto de reacciones finalice con la activación genómica a partir de un patrón de estímulo particular (48). Es así como se llega a la inserción de más receptores de membrana, maquinaria molecular para las reacciones intracelulares e isoformas moleculares, que contribuyen a la formación y mantenimiento del recuerdo a largo plazo.
Una quinasa de histonas es la proteína kinasa 1 mitógena y estrés activa, una molécula activada por la cascada de ERK y MAPK. Esta kinasa media la fosforilación mitogénica, y a su vez, inducida por estrés, de la histona 3, CREB, factor activador de transcripción 1, proteínas de alta movilidad entre otras (61). Por ello, esta enzima se ha propuesto como el suceso que precede a la remodelación de la cromatina, pues es el punto donde converge la actividad de los receptores y canales de membrana, así como segundos mensajeros y quinasas (61, 63).
LA ACCIÓN REGULADORA DE LAS GTPasas
Dentro de la familia Ras se encuentran las proteínas Rap GTPasas: Rap 1 y Rap 2 (64-66). Ambas moléculas se ubican pre y postsinápticamente (65), su activación se da por factores como Epac (activada por el AMPc) o por disminución de la actividad sináptica (65, 66). Rap 1 y Rap 2 participan en la depresión a largo plazo -mecanismo que revierte el fenómeno de potenciación a largo plazo-a través de varios mecanismos; es decir, que posee un efecto negativo sobre la PLP. Un mecanismo consiste en remover los receptores AMPA de la membrana postsináptica, especialmente la subunidad GluR2, promoviendo la internalización del receptor y su degradación, efecto que se verá reflejado en una menor transmisión glutamatérgica y menores potenciales excitatorios postsinápticos (66). Otro, mediante la actividad de Rap 2, es reducir la fosforilación de ERK y MAPK (64). Esos resultados demuestran la necesidad de actividad neuronal para el mantenimiento de la potenciación y se vislumbra cómo el sistema nervioso posee la facultad de moldearse en respuesta a su funcionamiento.
Aunque otro estudio muestra que, al inhibirse Rap 1, se liberan presinápticamente grandes cantidades de glutamato, lo que provoca una sobreactivación general. El problema es que también paralelamente se presenta activación de neuronas locales inhibitorias, y este suceso inhibitorio predomina sobre la excitación, elevando el umbral para las entradas sensoriales en el núcleo lateral de la amígdala, sobre todo a estímulos incondicionados débiles o señales complejas (65). Si cesa el mecanismo de modulación realizado por Rap 1 se crea una sobreactivación de otras vías moduladoras que terminan por ser más eficaces que la actividad excitatoria, y esto reduce la probabilidad de activación neuronal local; además, hace pensar de nuevo en el mecanismo de equilibrio entre activación e inhibición para que se presente la memoria. Su función consiste entonces en refinar las conexiones nerviosas de acuerdo con su frecuencia de actividad durante el desarrollo y vida de los animales, particularmente Rap2, influyendo en el crecimiento y retracción de dendritas y axones (64, 66).
UN NUEVO ROL DEL MIEDO EN LA MEMORIA
Desde hace algún tiempo se planteó que la memoria y el aprendizaje se ven reforzados por el miedo, lo que hizo que se comenzara a prestarle importancia como un suceso que favorece, entre otras cosas, el aprendizaje al provocar un refuerzo en la actividad neuronal (11). En otras palabras, la emoción ocasiona una sobreactivación de los circuitos neuronales, lo cual permite una intensa excitación entre ciertas sinapsis que se encuentren actuando de manera coherente, y con ello, desencadenando un fenómeno de PLP. Desde este punto de vista, el miedo sería algo así como una tetania fisiológica. Los cambios celulares consecuentes en cada célula implican la actuación de varios tipos de moléculas coordinadas que reforzarán algunas de sus sinapsis, y con ello, especificarán cuáles configuraciones neuronales se van a construir en momentos determinados. Allí es donde radica la causa de ese ser vital adaptable al ambiente que requiere transformar una memoria recién adquirida en un trazo durable (52) y enriquecer su comportamiento.
Tal relación (emoción-memoria) ha llevado a plantear la hipótesis de la modulación de la memoria. Ésta propone que pasada una experiencia de excitación emocional, se activan algunas estructuras límbicas como amígdala e hipocampo, y el eje hipotálamohipofisario-adrenal; y al interactuar, promueven el aprendizaje y la memoria (11). Aquí, el miedo no sólo es la emoción en la amenaza, sino que también entra como parte del suceso orgánico requerido para el fenómeno de aprendizaje y memoria, brindando una capacidad selectiva frente a cada evento que experimenta el organismo, permitiéndole recordar con mayor facilidad. Sin embargo, es muy probable que haya otras estructuras que sean indispensables, pues se debe recordar que el miedo es una configuración neuronal determinada, un estado fisiológico en ciertas condiciones (1, 2) que implica una amplia actividad dentro del sistema nervioso.
CONCLUSIÓN
Las células neuronales y gliales gozan de mecanismos moleculares que brindan la posibilidad de ir modificando en cierto grado la funcionalidad del sistema nervioso al modificar la actividad celular durante la vida del organismo. Esos hechos conllevan a construir individuos con características propias en sus conexiones sinápticas. Cada mecanismo se construye por una interacción de diversas moléculas con estructuras y funciones particulares. Las moléculas más importantes son los receptores de membranas, canales iónicos, proteínas G, enzimas, segundos mensajeros, factores de transcripción, histonas, ADN y ARN mensajero y de transferencia. Las propiedades de sus reacciones se encuentran definidas por los grupos funcionales que contienen, pudiendo ser porciones que se pierden o anexan en la reacción. Pueden, además, señalizar los sitios de proteólisis y activar la molécula. Una enzima puede poseer isoformas que revierten las acciones de su contraparte, al frenar las reacciones intracelulares de la adquisición y consolidación de la memoria y, por otro lado, servir para el mantenimiento del recuerdo. De ahí que exista un mecanismo regulador que invierta la activación inicial y pula sutilmente cada cambio celular, de manera que sean precisos y correspondientes a las circunstancias. Así mismo, las vías pueden conectarse en ciertos puntos, y hacer simultáneo cada proceso alterno y magnificar el resultado de las reacciones a escala celular.
El refinamiento da paso entonces a que el miedo pueda ser aprendido bien sea frente a un estímulo que antecede a una amenaza, o frente a un contexto en el que se asocien varios estímulos con una señal de peligro, para evitar de esta forma establecer una asociación generalizada del peligro con otras señales que no tengan relación y generen respuestas inapropiadas a la circunstancia. La variación a largo plazo es la conclusión de los eventos químicos, es el producto de la síntesis de nuevas moléculas a partir de la transcripción genética. Esas nuevas moléculas pueden ser enzimas, sustancias precursoras, sustratos, receptores, neurotransmisores y su incorporación refuerza la eficacia de las propiedades electroquímicas de comunicación neuronal. Otro aspecto importante, compatible con lo sugerido en escritos pasados sobre la neurobiología del miedo, es que la modulación y el equilibrio entre excitación e inhibición mantienen regulados los cambios intracelulares y los cambios en las conexiones del entramado celular; de tal forma, se evitan los extremos en la actividad que podrían conllevar a consecuencias negativas y originar trastornos neurológicos. Finalmente, el miedo puede modular el aprendizaje y la memoria por la excitación celular que provoca, dando inicio a esas vías bioquímicas; por lo tanto, ayuda a escoger qué se aprende y qué no.
RECOMENDACIONES
En estudios futuros es recomendable profundizar sobre los cambios funcionales en las células gliales, qué pasa en ellas al momento de la adquisición, reconsolidación y mantenimiento de la potenciación a largo plazo, integrando en un todo el fenómeno de la potenciación a largo plazo. Desglosando lo dicho un poco más, es entender cómo se hace más probable la excitación neuronal, mediante la producción de una mayor cantidad de potenciales excitatorios postsinápticos en respuesta a la transmisión proveniente de otras células. De igual forma, es importante la búsqueda de otras moléculas de diversos tipos e identificar sus acciones en el transporte de la señal extracelular al interior del citoplasma y el núcleo, ya que el conocimiento sobre el funcionamiento celular es la base para el desarrollo de soluciones a las enfermedades del sistema nervioso. Debe ser prioritario establecer la totalidad de las conexiones presentes en el miedo, y cómo éste es una configuración neuronal determinada.
AGRADECIMIENTOS
Agradecemos a Ana María González Aristizábal, Diseñadora Visual de la Universidad de Caldas.
REFERENCIAS
1. Sánchez-Ramírez JD, Uribe-Velásquez LF. Aspectos neurobiológicos implicados en el miedo animal. Biosalud 2009; 8:189-213. [ Links ]
2. Sánchez-Ramírez JD, Uribe-Velásquez LF. Neurobiología del miedo factor emocional y desencadenante motor visto desde sus bases moleculares, celulares y neuroanatómicas causantes. Monografía del Programa de Medicina Veterinaria y Zootecnia, Facultad de Ciencias Agropecuarias, Universidad de Caldas, Manizales; 2009. pp. 4-67. [ Links ]
3. Huang YY, Martin KC, Kandel ER. Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J Neurosci 2000; 20(17):6317-6325. [ Links ]
4. Ginas RR. El cerebro y el mito del yo. 1ra edición. Bogotá: Norma; 2003. pp. 181-201. [ Links ]
5. Álvarez RP, Biggs A, Chen G, Pine DS, Grillon C. Contextual fear conditioning in humans: Corticalhippocampal and amygdala contributions. J Neurosci 2008; 28(24):6211-6219. [ Links ]
6. Huang YY, Kandel ER. 5-Hydroxytryptamine induces a protein kinase A/Mitogen-activated protein kinase-mediated and macromolecular synthesis-dependent late phase of long-term potentiation in the amygdala. J Neurosci 2007; 27(12):3111-3119. [ Links ]
7. Kalin NH. Neurobiología del miedo. Sci Am 1993; 268(5):94-101. [ Links ]
8. Carlson NR. Fisiología de la conducta. 6ta edición. Barcelona: Ariel Neurociencia; 1999. pp. 405-430. [ Links ]
9. Schafe GE, LeDoux JE. Memory consolidation of auditory Pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci 2000; 20:1-5. [ Links ]
10. Cohen JD, Castro-Alamancos MA. Early sensory pathways for detection of fearful conditioned stimuli: Tectal and thalamic relays. J Neurosci 2007; 27(29):7762-7776. [ Links ]
11. LaBar KS. Beyond fear emotional memory mechanisms in the human brain. Curr Dir Psychol Sci 2007; 16(4):173-177. [ Links ]
12. Kishioka A, Fukushima F, Ito T, Kataoka H, Mori H, Ikeda T, et al. A novel form of memory for auditory fear conditioning at a low-intensity unconditioned stimulus. PLoS One 2009; 4(1):e4157. [ Links ]
13. Christensen JW, Rundgren M. Predator odour per se does not frighten domestic horses. J Applanim 2007; 112(1-2):136-145. [ Links ]
14. Guimarães-Costa R, Guimarães-Costa MB, Pippa-Gadioli L, Weltson A, Ubiali WA, Paschoalin-Maurin T, et al. Innate defensive behaviour and panic-like reactions evoked by rodents during aggressive encounters with Brazilian constrictor snakes in a complex labyrinth: Behavioural validation of a new model to study affective and agonistic reactions in a prey versus predator paradigm. J Neurosci Methods 2007; 165:25-37. [ Links ]
15. Bass SLS, Gerlai R. Zebrafish (Danio rerio) responds differentially to stimulus fish: The effects of sympatric and allopatric predators and harmless fish. Behav Brain Res 2008; 186(1):107-117. [ Links ]
16. Takahashi LK, Chan MM, Pilar ML. Predator odor fear conditioning: Current perspectives and new directions. Neurosci Biobehav Rev 2008; 32(7):1218-1227. [ Links ]
17. Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J Neurosci 2000; 20(21):8177-8187. [ Links ]
18. Apergis-Schoute AM, De.biec J, Doyére V, LeDoux JE, Schafe GE. Auditory fear conditioning and long-term potentiation in the lateral amygdala require ERK/MAP kinase signaling in the auditory thalamus: A role for presynaptic plasticity in the fear system. J Neurosci 2005; 25:5730-5739. [ Links ]
19. Lewis MC, Gould TJ. Signal transduction mechanisms within the entorhinal cortex that support latent inhibition of cued fear conditioning. Neurobiol Learn Mem 2007; 88(3):359-368. [ Links ]
20. Schafe GE, Swank MW, Rodrigues SM, De.biec J, Doyère V. Phosphorylation of ERK/MAP kinase is required for long-term potentiation in anatomically restricted regions of the lateral amygdala in vivo. Learn Mem 2008; 15:55-62. [ Links ]
21. Fischer A, Radulovic M, Schrick C, Sananbenesi F, Godovac-Zimmermann J, Radulovic J. Hippocampal Mek/Erk signaling mediates extinction of contextual freezing behavior. Neurobiol Learn Mem 2007; 87(1):149-158. [ Links ]
22. Santini E, Quirk GJ, Porter JT. Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbic neurons. J Neurosci 2008; 28(15):4028-4036. [ Links ]
23. Schafe GE, Nadel NV, Sullivan GM, Harris A, LeDoux JE. Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn Mem 1999; 6:97-110. [ Links ]
24. Kiyama Y, Manabe T, Sakimura K, Kawakami F, Mori H, Mishina M. Increased thresholds for long-term potentiation and contextual learning in mice lacking the NMDA-type glutamate receptor  1 subunit. J Neurosci. 1998; 18(17):6704-6712. [ Links ]
1 subunit. J Neurosci. 1998; 18(17):6704-6712. [ Links ]
25. Weisskopf MG, Bauer EP, LeDoux JE. L-type voltage-gated calcium channels mediate NMDA independent associative long-term potentiation at thalamic input synapses to the amygdala. J Neurosci 1999; 19(23):10512-10519. [ Links ]
26. Macrì S, Pasquali P, Bonsignore LT, Pieretti S, Cirulli F, Chiarotti F, et al. Moderate neonatal stress decreases within-group variation in behavioral, immune and HPA responses in adult mice. PLoS One 2007; 10:e1015. [ Links ]
27. Wilensky AE, Schafe GE, LeDoux JE. The amygdala modulates memory consolidation of fear-motivated inhibitory avoidance learning but not classical fear conditioning. J Neurosci 2000; 20:7059-7066. [ Links ]
28. Chourbaji S, Hellweg R, Brandis D, Zörner B, Zacher C, Lang UE, et al. Mice with reduced brain-derived neurotrophic factor expression show decreased choline acetyltransferase activity, but regular brain monoamine levels and unaltered emotional behavior. Brain Res Brain Mol Res 2004; 121:28-36. [ Links ]
29. Rodrigues SM, Farb CR, Bauer EP, LeDoux JE, Schafe GE. Pavlovian fear conditioning regulates Thr286 autophosphorylation of Ca2+/Calmodulin-dependent protein kinase II at lateral amygdala synapses. J Neurosci 2004; 24:3281-3288. [ Links ]
30. Runyan JD, Moore AN, Dash PK. A role for prefrontal cortex in memory storage for trace fear conditioning. J Neurosci 2004; 24:1288-1295. [ Links ]
31. Lubin FD, Sweatt JD. The I B kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron 2007; 55:942-957. [ Links ]
B kinase regulates chromatin structure during reconsolidation of conditioned fear memories. Neuron 2007; 55:942-957. [ Links ]
32. Fails WA, Miserendino MJD, Davis M. Extinction of fear-potentiated startle: Blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci 1992; 12(3):854-963. [ Links ]
33. Blum S, Runyan JD, Dash PK. Inhibition of prefrontal protein synthesis following recall does not disrupt memory for trace fear conditioning. BMC Neurosci 2006; 7(67):1-10. [ Links ]
34. Duvarci S, Nader K, LeDoux JE. de novo mRNA synthesis is required for both consolidation and reconsolidation of fear memories in the amygdala. Learn Mem 2008; 15(10):747-755. [ Links ]
35. Ouyang M, Zhang L, Zhu JJ, Schwede F, Thomas SA. Epac signaling is required for hippocampus dependent memory retrieval. PNAS 2008; 105(33):11993-11997. [ Links ]
36. Pistell PJ, Falls WA. Extended fear conditioning reveals a role for both N-methyl-D-aspartic acid and non-N-methyl-D-aspartic acid receptors in the amygdala in the acquisition of conditioned fear. Neurosci 2008; 155(4):1011-20. [ Links ]
37. Bourtchouladze R, Abel T, Berman N, Gordon R, Lapidus K, Kandel ER. Different training procedures recruit either one or two critical periods for contextual memory consolidation, each of which requires protein synthesis and PKA. Learn Mem 1998; 5:365-374. [ Links ]
38. Lu KT, Walker DL, Davis M. Mitogen-activated protein kinase cascade in the basolateral nucleus of amygdala is involved in extinction of fear-potentiated startle. J Neurosci 2001; 21:1-5. [ Links ]
39. Lin CH, Yeh SH, Lu HY, Gean PW. The similarities and diversities of signal pathways leading to consolidation of conditioning and consolidation of extinction of fear memory. J Neurosci 2003; 23(23):8310-8317. [ Links ]
40. De.biec J, Doyère V, Nader K, LeDoux JE. Directly reactivated, but not indirectly reactivated, memories undergo reconsolidation in the amygdala. PNAS 2006; 103:3428-3433. [ Links ]
41. Palucha A, Pilc A. Metabotropic glutamate receptor ligands as possible anxiolytic and antidepressant drugs. Pharmacol Ther 2007; 115:116-147. [ Links ]
42. Balschun D, Wolfer DP, Gass P, Mantamadiotis T, Welzl H, Schütz G, et al. Does cAMP response elementbinding protein have a pivotal role in hippocampal synaptic plasticity and hippocampus-dependent memory? J Neurosci 2003; 23(15):6304-6314. [ Links ]
43. Korzus E, Rosenfeld MG, Mayford M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 2004; 42:961-972. [ Links ]
44. Isiegas C, Park A, Kandel ER, Abel T, Lattal KM. Transgenic Inhibition of Neuronal Protein Kinase A Activity Facilitates Fear Extinction. J Neurosci 2006; 26(49):12700-12707. [ Links ]
45. Isiegas C, McDonough C, Huang T, Havekes R, Fabian S, Wu LJ, et al. A novel conditional genetic system reveals that increasing neuronal cAMP enhances memory and retrieval. J Neurosci 2008; 28(24):6220-6230. [ Links ]
46. Kojima N, Borlikova G, Sakamoto T, Yamada K, Ikeda T, Itohara S, et al. Inducible cAMP early repressor acts as a negative regulator for kindling epileptogenesis and long-term fear memory. J Neurosci 2008; 28(25):6459-6472. [ Links ]
47. Kelly MP, Cheung YF, Favilla C, Siegel SJ, Kanes SJ, Houslay MD, et al. Constitutive activation of the G-protein subunit G s within forebrain neurons causes PKA-dependent alterations in fear conditioning and cortical Arc mRNA expression. Learn Mem 2008; 15:75-83. [ Links ]
s within forebrain neurons causes PKA-dependent alterations in fear conditioning and cortical Arc mRNA expression. Learn Mem 2008; 15:75-83. [ Links ]
48. Wood MA, Hawk JD, Abel T. Combinatorial chromatin modifications and memory storage: A code for memory? Learn Mem 2006; 13:241-244. [ Links ]
49. Maren S, Fanselow MS. Synaptic plasticity in the basolateral induced by amygdala hippocampal formation stimulation in viva. J Neurosci 1995; 15(11):7546-7564. [ Links ]
50. Bauer EP, Schafe GE, LeDoux JE. NMDA Receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci 2002; 22(12):5239-5249. [ Links ]
51. Vaas R. Neurobiología del miedo. Mente y cerebro 2002; 1:56-63. [ Links ]
52. Kalisch R, Holt B, Petrovic P, De Martino B, Klöppel S, Büchel C, et al. The NMDA agonist D-cycloserine facilitates fear memory consolidation in humans. Cereb Cortex 2009; 19:187-196. [ Links ]
53. Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AMM, Lombardi TL, et al. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem 2005; 12:111-119. [ Links ]
54. English JD, Sweatt JD. Activation of p42 mitogenactivated protein kinase in hippocampal long term potentiation. J Biol Chem 1996; 271:24329-24332. [ Links ]
55. Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, et al. Histone Deacetylase inhibitors enhance memory and synaptic plasticity via CREB: CBP-Dependent transcriptional activation. J Neurosci 2007; 27(23):6128-6140. [ Links ]
56. Ryu J, Futai K, Feliu M, Weinberg R, Sheng M. Constitutively active Rap2 transgenic mice display fewer dendritic spines, reduced extracellular signal-regulated kinase signaling, enhanced long-term depression, and impaired spatial learning and fear extinction. J Neurosci 2008; 28(33):8178-8188. [ Links ]
57. Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: Requirement for GluR1 and PDZ domain interaction. Science 2000; 287:2262-2267. [ Links ]
58. Gresack JE, Schafe GE, Orr PT, Frick KM. Sex differences in contextual fear conditioning are associated with differential ventral hippocampal extracellular signal-regulated kinase activation. Neuroscience 2009; 159(2):451-67. [ Links ]
59. Schafe GE, Doyère V, LeDoux JE. Tracking the fear engram: The lateral amygdala is an essential locus of fear memory storage. J Neurosci 2005; 25(43):10010-10015. [ Links ]
60. Hebert AE, Dash PK. Plasticity in the entorhinal cortex suppresses memory for contextual fear. J Neurosci 2004; 24(45):10111-10116. [ Links ]
61. Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem 2004; 279(39):40545-40559. [ Links ]
62. Chwang WB, Arthur JS, Schumacher A, Sweatt JD. The nuclear kinase mitogen-and stress-activated protein kinase 1 regulates hippocampal chromatin remodeling in memory formation. J Neurosci 2007; 27(46):12732-12742. [ Links ]
63. Pan BX, Vautier F, Ito W, Bolshakov VY, Morozov A. Enhanced cortico-amygdala efficacy and suppressed fear in absence of Rap1. J Neurosci 2008; 28(9):2089-2098. [ Links ]
64. Rodrigues SM, Schafe GE, LeDoux JE. Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J Neurosci 2001; 21(17):6889-6896. [ Links ]
65. Fu Z, Lee SH, Simonetta A, Hansen J, Sheng M, Pak DTS. Differential roles of Rap1 and Rap2 small GTPases in neurite retraction and synapse elimination in hippocampal spiny neurons. J Neurochem 2007; 100(1):118-131. [ Links ]
66. Serrano P, Friedman EL, Kenney J, Taubenfeld SM, Zimmerman JM, Hanna J, et al. PKM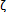 Maintains spatial, instrumental, and classically conditioned long-term memories. PLoS Biology 2008; 6:2698-2706. [ Links ]
Maintains spatial, instrumental, and classically conditioned long-term memories. PLoS Biology 2008; 6:2698-2706. [ Links ]

