










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Ciencias de la Salud
versión impresa ISSN 1692-7273versión On-line ISSN 2145-4507
Rev. Cienc. Salud v.4 n.1 Bogotá ene./jun. 2006
Fenotipo y genotipo de once pacientes con Ataxia de Friedreich
Friedreich's Ataxia: Phenotype and Genotype in Eleven Patients
Elizabeth Vargas, Victoria Eugenia Villegas, Olga Lucía Pedraza, Clemencia Durán, Juan Carlos Prieto*
* Elizabeth Vargas: Estudiante de Bacteriología, Universidad Javeriana, Bogotá D. C.
Victoria Eugenia Villegas: Bióloga MSc, Universidad Javeriana, Bogotá D. C.
Olga Lucía Pedraza: Médica Cirujana, Universidad Javeriana, Bogotá D. C.
Clemencia Durán: Bióloga MSc, Universidad Javeriana, Bogotá D. C.
Juan Carlos Prieto: Médico Genetista, Universidad Javeriana, Bogotá D. C. Este trabajo se realizó con el apoyo del Instituto de Genética de la Universidad Javeriana. Correspondencia: Doctor Juan Carlos Prieto; Carrera 7 No 40-62. Edificio 32. Instituto de Genética Humana. Universidad Javeriana. Bogotá. E-mail: jcprieto@javeriana.edu.co
Recibido: marzo 2 de 2006 Aceptado: marzo 21 de 2006
Resumen
Introducción: La ataxia de Friedreich (FRDA) es una enfermedad autosómica recesiva debida a una mutación en el gen X25. Dicho gen está localizado en el cromosoma 9 y codifica para la proteína frataxina. La enfermedad es causada por la repetición del trinucleótido GAA. En individuos normales la secuencia GAA se encuentra repetida entre siete y veintidós veces, mientras que, en pacientes con ataxia de Friedreich GAA puede estar repetida cientos o miles de veces.
Objetivos: Evaluar si existe correlación entre el tamaño de la expansión, la edad de inicio de FRDA y su severidad en la muestra seleccionada.
Métodos: - Se estudiaron once pacientes con fenotipo típico de ataxia de Friedreich. El análisis molecular por PCR determinó la expansión del trinucleótido GAA. Se analizó la correlación entre la edad de inicio de FRDA y su progresión con el número de repeticiones GAA.
Resultados y conclusiones: - El análisis molecular por PCR mostró ocho pacientes homocigotos para la expansión, y tres negativos. El promedio del tamaño de las expansiones en los alelos es 622±5 con un promedio correspondiente de la edad inicio de FRDA 13±8. Para el tamaño de la muestra no se observó una correlación estadística significativa entre la edad de inicio de la enfermedad y el número de repeticiones, pero sí una tendencia a correlacionarse de forma inversa (p<0.11). El diagnóstico molecular de FRDA sumado a la comprensión de su fisiología y a la utilización de los criterios de inclusión de Harding, constituye un paso importante en el logro de un tratamiento óptimo de la enfermedad.
Palabras Claves: Friedreich ataxia, repeticiones GAA, alelos de riesgo
Abstract
Introduction: - Friedreich's ataxia is an autosomal recessive disease due to a mutation in gene X25. This gene codes for frataxin and it is located on chromosome 9. The disease is caused by a triplet particular sequence of bases (GAA). Normally, the GAA sequence is repeated 7 to 22 times, but in people with Friedreich's ataxia, it can be repeated hundreds or even over thousand times.
Objectives: To determine if there is a correlation between clinical and molecular findings in our FRDA patients.
Methods: Eleven patients with the typical Friedreich´s ataxia phenotype were studied by PCR we determined the size of the GAA expansions, and analyzed the correlation of age at onset and rate of disease progression with the number of GAA repetitions.
Results and conclusions: Molecular analysis by PCR showed eight homozygous patients for the expansion and three negative. The average of the size of the expansions in the allele was of 622±5 with an average in the age of beginning of 13±8. For the sample size, there was no significant statistical correlation between the age of beginning of the disease and the number of repetitions, although there was like an inverse correlation.
Besides understanding of FRDA physiology and the Harding clinical inclusion criteria, molecular diagnosis is an important step in the achievement of an optimal therapeutic treatment.
Key words: Friedreich ataxia, GAA repeats, risk allele.
INTRODUCCIÓN
La ataxia de Friedreich (FRDA) está catalogada como una enfermedad neurodegenerativa, siendo la más común de las ataxias. Antes de la disponibilidad de diagnóstico molecular, se estimaba que la FRDA afectaba aproximadamente 1:50 000 individuos con una prevalencia de portadores de 1:110. Los más recientes estudios están basados en datos moleculares que hacen pensar en una prevalencia mayor. Según un estudio realizado en Alemania, en una muestra de 178 individuos sanos fue hallada una prevalencia de portadores de 1/60 - 1/90 (1).
Las características físicas de los pacientes con ataxia de Friedreich son: ataxia de la marcha, arreflexia en los cuatro miembros asociada con la disartria, pérdida distal tanto de la sensibilidad vibratoria como de la propioceptiva, con signos piramidales. El inicio de la sintomatología ocurrente antes de veinte años y las complicaciones asociadas con la patología incluyen cardiomiopatia, atrofia óptica, hipoacusia y diabetes mellitus. El criterio de diagnóstico en pacientes con FRDA está basado en las manifestaciones clínicas establecidas por Harding en 1981 (2-4).
Han sido descritas dos variantes de FRDA: la variante FARR o ataxia de Friedreich con reflejos conservados, y la forma LOFA o ataxia de Friedreich de inicio tardío a partir de los veinticinco años de edad (5-7).
El gen responsable de FRDA fue mapeado en el cromosoma 9 por Chamberlain en 1988. Consta de siete exones 1, 2, 3, 4, 5a, que transcriben a una proteína de 210 aminoácidos. El 5b funciona como splicing alternativo en el 5a, mientras que el exón 6 no codifica. Este gen fue llamado x25 o frataxina ya que codifica para la proteína del mismo nombre (1-8).
El papel de la frataxina no se conoce completamente. La mutación más común en FRDA es la expansión de la repetición del trinucleótido GAA en el intrón 1 del gen X25. Los individuos sanos se caracterizan por tener entre siete y veintidós repeticiones GAA, mientras que los pacientes de FRDA tienen entre doscientas y novecientas repeticiones. (8-9). Experimentos in vivo e in vitro indican que la repetición GAA interfiere con la transcripción. La base molecular de este resultado corresponde a la conformación de una molécula de DNA con una estructura de triple hélice (α-DNA). Este fenómeno ha sido llamado Sticky DNA, donde hay una asociación de dos purinas y una pirimidina (R-R-Y). Esta última base in fluye negativamente en el proceso de enrollamiento del DNA, lo que provoca una supresión en la expresión del gen y, por consiguiente, una reducción en los niveles de proteína (10-11).
La frataxina, proteína localizada en la mitocondria, es clave para el control del metabolismo del hierro y de los radicales libres. Los pacientes con FRDA sólo tienen una deficiencia parcial de la proteína y no una carencia completa, lo que hace que la deficiencia esté asociada con una disfunción de la mitocondria, compatible con las características clínico-patológicas de la enfermedad. Estas anomalías incluyen deficiencia de las enzimas piruvato carboxilasa y lipoamida deshidrogenasa, pertenecientes a la cadena respiratoria (4).
Es probable que el papel primario de la frataxina esté relacionado con la biosíntesis de Fe/S de las proteínas, mecanismo que llevaría a la reducción de la energía celular y al aumento de la producción de radicales libres. Esto influiría en las proteínas críticas de la cadena respiratoria y en las subunidades de los complejos I, II, y III. Independientemente del mecanismo mitocondrial involucrado, la acumulación férrica puede contribuir a la producción de las especies de oxígeno reactivo (ROS) y a la generación de radicales libres a través de la reacción de Fenton. El radical OH -en presencia de exceso de hierropermite que, tanto proteínas como carbohidratos y lípidos reaccionen con el DNA y tanto, alteren su estabilidad (1, 4, 10, 12).
Este eslabón entre la patología y el estado prooxidante, hace de la FRDA un excelente modelo de enfermedad neurodegenerativa, en donde los radicales reactivos y la tensión oxidante contribuyen a la progresión de la enfermedad (13).
MATERIALES Y MÉTODOS
Fueron analizados once pacientes remitidos por la Unidad de Neurología del Hospital Universitario de San Ignacio, debido a ataxia progresiva y a clínica compatible con la enfermedad de FRDA.
Todos los pacientes fueron examinados clínicamente por el mismo neurólogo, según el criterio de diagnóstico de Harding (1981). Del total de pacientes, ocho fueron diagnosticados con Ataxia de Friedreich clásica y tres más con la variante FARR.
Análisis en la repetición GAA
Las muestras de sangre fueron tomadas con anticoagulante EDTA y extraídas por el método de Fenol-Cloroformo. El análisis de la repetición GAA del primer intrón del gen X25 fue determinado con los primers o cebadores diseñados por Campuzano en 1996: GAAF5 ´GGGATTGGTTGCCAGTGCTTAAAAGTTAG3 ´ y GAAR5 ´GATCTA AGGACCATCATGGCCACACTTGCC3. Estos cebadores actúan en la expansión de la tripleta GAA y generan productos 457 + 3n pb (n = numero de tripletas GAA) (9). Para la amplificación se utilizó el Kit de Elongasa (Gibco) bajo las siguientes condiciones: desnaturalización inicial de 94 °C por tres minutos, seguida de diez ciclos a 94 °C por treinta segundos, 59 °C por treinta segundos, 68 °C por tres minutos, y veinte ciclos a 94 °C por treinta segundos, 62 °C por treinta segundos y, finalmente, a 68 °C por tres minutos, con un incremento de veinte segundos en cada ciclo. Los productos de PCR fueron separados mediante electroforesis en gel de agarosa al 1.5% y visualizados con bromuro de etidio en cámara con radiación UV. El tamaño de los alelos de cada paciente fue determinado mediante comparación del marcador de peso 1Kb.
Como prueba estadística se utilizó el coeficiente de correlación de Pearson para dos variables predictorias: el tamaño de los alelos, y edad de inicio y severidad de FRDA.
RESULTADOS Y DISCUSIÓN
La determinación del número de repeticio nes GAA por medio de la PCR permite confirmar el diagnóstico de FRDA (Ver Figura 1). En este estudio fueron diagnosticados clínicamente ocho pacientes con Ataxia de Friedreich clásica y tres con la variante FARR. En el 88% de los pacientes se encontró la repetición GAA y en el 12% restante la expansión fue negativa.
La Tabla 1 muestra la comparación clínica y genética en los pacientes afectados: en un 46% se encontró Ataxia clásica de FRDA, en el 27% la variante FARR, y en el 27% ausencia de expansión de la repetición GAA. Este último resultado puede deberse a una fenocopia de FRDA o a una deficiencia de vitamina E (Ver Figura 2).
Las dificultades para reconocer las diferentes formas clínicas de ataxia hereditaria han existido desde el mismo momento en que diferentes grupos de investigadores iniciaron el estudio de las mismas. Por eso, es de gran importancia efectuar el estudio molecular, especialmente en los casos de FRDA y de déficit de vitamina E (14).
La relación genotipo-fenotipo fue estudiada en ocho pacientes homocigotos a la expansión (no hubo ningún paciente heterocigoto). Al alelo corto se le llamó GAA1 y al largo, GAA2. La media del tamaño de la expansión en los alelos fue 622±5 repeticiones, con un rango de 411-848 (DS = 140±2). En estos casos, la edad de inicio tiene una media de 13±8 (rango 5-20). En razón del pequeño tamaño de muestra, no se observó una correlación estadística significativa entre la edad de inicio de la enfermedad y el número de repeticiones, aunque sí fue evidente una tendencia a correlacionarse de forma inversa. A mayor número de repeticiones GAA, fue menor la edad de inicio de FRDA (p<0.11) (Ver Figura 3). Este resultado es similar a los hallazgos de Dürr y sus compañeros de investigación, quienes en 1996 reportaron que el tamaño de la expansión de GAA menor presenta una correlación importante tanto con la edad de inicio de la enfermedad como con su severidad. El número de repeticiones en el alelo de la expansión menor corresponde aproximadamente al 50% de la variabilidad en la edad de inicio de FRDA, comparado con menos del 20% para el alelo con la expansión más grande y 30% de la variación debida a factores desconocidos (2).
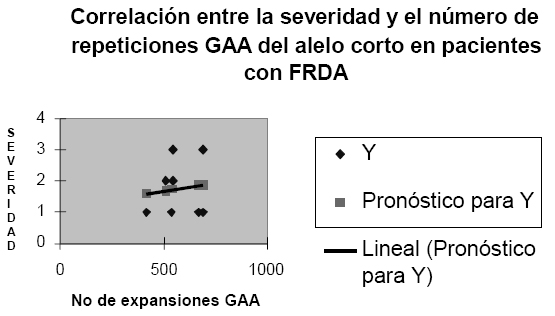
Entre el número de repeticiones GAA y el grado de severidad de la enfermedad se presentó también una tendencia a correlacionarse de manera inversa (Ver Figura 4). Teniendo en cuenta el carácter recesivo de FRDA, se podría suponer - según Filla et al (15)- que la severidad de la enfermedad se debe a una inusual inhibición de la transcripción del gen. Por consiguiente, no es sorprendente que la severidad de la enfermedad tenga una correlación con el alelo más corto en número de repeticiones GAA. Esta hipótesis es también reportada por Prieto (16) quien sugirió que las expansiones no inhiben totalmente la transcripción o la maduración del RNAm, sino que permiten una expresión residual de la proteína.
Tres pacientes con la variante FARR fueron diagnosticados homocigotos a la expansión de la repetición GAA, con tamaños de alelos que no diferían del genotipo de FRDA clásico. Sin embargo, la edad de inicio de la enfermedad para el grupo FARR fue más avanzada que para el grupo FRDA, hallazgo contrario al reportado por Lamont et al (6) quienes no encontraron una diferencia significativa entre las edades de inicio de los dos grupos.
En resumen, el análisis molecular efectuado en este estudio muestra la expansión de la repetición GAA descrita por Campuzano et al (9). En los ocho pacientes homocigotos en los que se confirmó que el número de repeticiones GAA, la edad de inicio y la severidad de la enfermedad presentaban tendencia a correlacionarse de for ma inversa, la causa principal de la FRDA fue la expansión de la repetición GAA.
En este orden de ideas, la posibilidad de avanzar en el tratamiento terapéutico de la FRDA depende no sólo de una mejor comprensión de la fisiopatología de la enfermedad que tome en cuenta los criterios clínicos de inclusión de Harding, sino también de un diagnóstico molecular fundamentado en la determinación del número de repeticiones del trinucleótido GAA.
AGRADECIMIENTOS
Este estudio fue posible gracias al apoyo financiero del Instituto de Genética Humana de la Pontificia Universidad Javeriana, Bogotá D. C. Agradecemos a los pacientes y a la Unidad de Neurología del Departamento de Neurociencias del Hospital San Ignacio, Bogotá D. C.
Tabla 1. Comparación clínica y genética en los pacientes con Friedreich
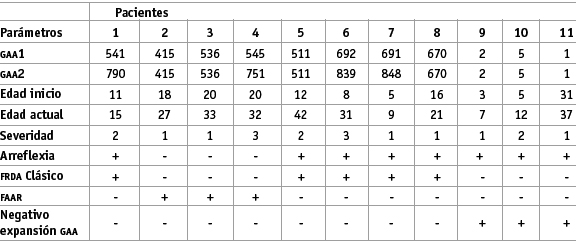
Figura 1. Gel de agarosa 1.5%. Amplificación de los productos PCR del intrón 1 del gen X25, donde se encuentra la expansión de la repetición GAA del mismo paciente en dos ensayos diferentes. Se incluye la estandarización de la técnica.
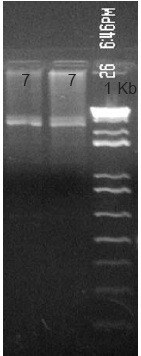
Figura 2. Gel de agarosa 1.5%. Amplificación PCR de pacientes diagnosticados clínicamente como FRDA y confirmados molecularmente como negativos para la expansión de la repetición GAA.
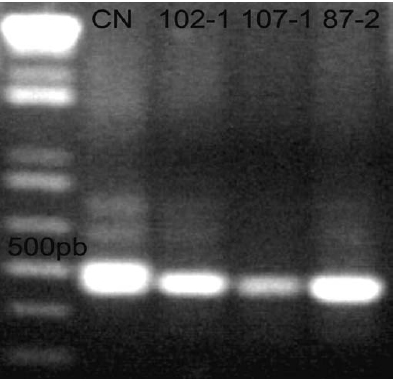
Figura 3 Correlación entre la edad de inicio y el número de repeticiones GAA en el alelo corto en pacientes con FRDA
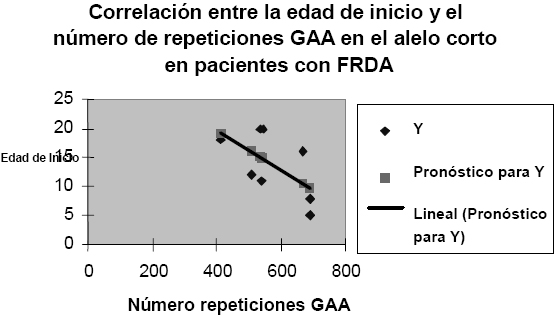
Figura 4 Correlación entre la severidad y el número de repeticiones GAA del alelo corto en pacientes con FRDA
BIBLIOGRAFÍA
1. Delatycki MB, Williamson R, Forrest S. Friedreich Ataxia: an overview. J Med Genet 2000; 37:1-8. [ Links ]
2. Dürr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C et al. Clinical and genetic abnormalities in patients with Friedreich´s Ataxia. N Engl J Med 1996; 335:1169-1175. [ Links ]
3. Leone M, Rocca WA, Rosso MG, Mantel N, Schoenberg BS, Schiffer D. Friedreich's disease: Survival analysis in an italian population. Neurology 1988; 38: 1433-1438. [ Links ]
4. Massimo P. Molecular Pathogenesis of Friedreich Ataxia. Arch Neurol 1999; 56: 1201-207. [ Links ]
5. Harding AE. Early onset cerebellar ataxia with retained tendon reflexes: a clinical and genetic study of a disorder distinct from Friedreich's ataxia. J Neurol Neurosurg Psychiatr 1981; 44: 503–508. [ Links ]
6. Lamont PJ, Davis MB, Wood NW. Identification and sizing of the GAA trinucleotide repeat expansion of Friedreich ataxia in 56 patients. Brain 1997; 120: 673-680. [ Links ]
7. Palau F, De Michele G, Vilchez JJ, Pandolfo M, Monros E, Cocozza S et al. Early on - set ataxia with cardiomyopathy and retained tendon reflexes maps to the Friedreich's ataxia locus on chromosome 9q. Ann Neurol 1995; 37: 359–362. [ Links ]
8. Schols L, Amoiridis G, Przuntek H, Frank G, Epplen JT, Epplen C. Friedreich's Ataxia Revision of the phenotype according to molecular genetics. Brain 1997; 120: 2131-2140. [ Links ]
9. Campuzano V, Montermini L, Moltó MD, Pianese L, Cossée M, Cavalcanti F et al. Friedreich ataxia: autosomal recessive disease caused by an intronic GAA repeat expansion. Science 1996; 271:1423-1430. [ Links ]
10. Puccio H, Koening M. Recent advances in the molecular pathogenesis of Friedreich Ataxia. Hum Mol Genet 2000; 9:887-892. [ Links ]
11. Sakamoto N, Chastain PD, Parniewski P, Ohshima K, Pandolfo M, Griffith JD et al. Sticky DNA: selfassociation properties of long GAA-TTC repeats in R-R-Y triplex structures from Friedreich´s ataxia. Mol Cell 1999; 3: 465-475. [ Links ]
12. Voncken M, Ioannoun P, Delaticky MB. Friedreich ataxia- update on pathogenesis and possible therapies. Neurogenetics 2004; 5: 1-8. [ Links ]
13. Jauslin ML, Wirth T, Meier T, Schoumacher F. A cellular model for Friedreich Ataxia reveals small - molecule glutathione peroxidase mimetics as novel treatment strategy. Hum Mol Genet 2002; 11:3055-3063. [ Links ]
14. Pedraza OL, Prieto JC, Casasbuenas OL, Espinosa E. Identificación clínica de las ataxias hereditarias. Acta Neurol Colomb 1996; 12: 132-139. [ Links ]
15. Filla A, De Michele G, Cavalcanti F, Pianese L, Monticelli A, Campanella G et al. The relationship between Trinucleotide (GAA) Repeat Length and Clinical Features in Friedreich Ataxia. Am J Hum Genet 1996; 59: 554-560. [ Links ]
16. Prieto JC, Pedraza OL, Gómez M, Durán C. Análisis molecular de la ataxia de Friedreich en Colombia. Acta Neurol Colomb 2004; 20: 7-12. [ Links ]

