










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Ciencias de la Salud
Print version ISSN 1692-7273On-line version ISSN 2145-4507
Rev. Cienc. Salud vol.6 no.3 Bogotá Sep./Dec. 2008
Enfermedad de Creutzfeldt-Jakob esporádica: estudio clínico, patológico y molecular de un caso
Sporadic Creutzfeldt-Jakob disease: Clinical, pathological and molecular study
Victoria Eugenia Villegas,1 Fernando Velandia,2 Cesar Payán3
1. Biol. MSc. Profesor asistente facultad de ciencias, Universidad del Rosario. Correo electrónico: victoria.villegasga@urosario.edu.co
2. Esp. Patólogo, neuropatólogo. Profesor principal. Miembro del Grupo de Investigación en Neurociencias (NEUROS), Universidad el Rosario. Correo electrónico: fervelah@gmail.com.
3. M.D. MSc. Profesor asistente facultad de ciencias. Universidad del Rosario. Correo electrónico: payangomezdelar@gmail.com
Recibido: 3 de octubre de 2008 Aceptado: 1 de noviembre de 2008
Resumen
Generalidades. Las encefalopatías espongiformes transmisibles son enfermedades neurodegenerativas ocasionadas por la acumulación anormal de una variante mal plegada de la proteína priónica, lo cual induce la formación de conglomerados proteicos resistentes a la degradación. Además, son responsables de la disfunción sináptica, daño neuronal y de la sintomatología clásica acompañante. Esta proteína de membrana es codificada por el exón 2 del gen PRNP, ubicado en el brazo corto del cromosoma 20 y parece estar involucrada en la trasmisión sináptica, la transducción de señales, la actividad antioxidante de la superoxidodismutasa, neuroplasticidad y sobrevida celular. Un polimorfismo en el codón 129 se asocia con una susceptibilidad diferencial a la enfermedad Creutzfeldt-Jakob esporádica.
Objetivo. Estudio clínico, patológico y molecular de un caso de una mujer de 58 años con diagnóstico de enfermedad de Creutzfeldt- Jakob esporádica.
Métodos y resultados. Se presenta el caso de una mujer en quien aparece un trastorno depresivo del afecto con demencia progresiva y sintomatología general. Al final de la enfermedad, el cuadro progresó a un déficit neurológico focalizado en el área visual. La RMN mostró hiperintensidades inespecíficas córtico-subcorticales en el núcleo estriado; en el EEG se encontró pérdida de ritmos de fondo, patrón de descargas periódicas generalizadas y complejos trifásicos; en la biopsia cerebral postmorten se evidenció pérdida severa de la población neuronal en todas las capas, vacuolas en el neuropil, en el soma neuronal y en la glía. El análisis de secuencia del gen PRNP, a partir de extracción de DNA de sangre periférica, identificó homocigosis para metionina en el codón 129. La paciente fallece a los 3 meses del inicio de la sintomatología.
Conclusión. Por epidemiología, curso clínico y exámenes paraclínicos se confirma el diagnóstico de enfermedad de Creutzfeldt- Jakob esporádica.
La determinación del genotipo para los polimorfismos de riesgo se convierte en una herramienta útil para complementar por medios moleculares el diagnóstico y para profundizar la comprensión de la fisiopatología de la enfermedad de Creutzfeldt-Jakob, tanto para formas esporádicas como para la nueva variante.
Palabras claves: enfermedad de Creutzfeldt- Jakob, prión, polimorfismos.
Abstract
Background: Transmissible spongiform encephalopathies are neurodegenerative diseases caused by abnormal accumulation of pathogenic isoform the prion protein, which induces the formation of conglomerates protein resistant to degradation. They are also responsible for synaptic dysfunction, neuronal damage and the classic symptoms of disease. This membrane protein is encoded by exon 2 of the gene PRNP, located on the short arm of chromosome 20 and appears to be involved in synaptic transmission, signal transduction, the antioxidant activity of the superoxid dismutasa, neuroplasticity and cell survival. One polymorphism at codon 129 is associated with differential susceptibility to disease sporadic Creutzfeldt-Jakob disease.
Aim: Clinical, pathological and molecular report on an 58 year-old woman with pathological diagnosis of Creutzfeldt-Jakob sporadic disease.
Methods and results. The clinic course appears with a behavior depressive disorder with progressive dementia and symptoms. At the end of the disease, the scenario progressed to a neurological deficit focused on the visual area. The MRI showed nonspecific hyperintensity in cortiço-subcortical nucleus in the striatum, the EEG showed patterns of recurrent generalized discharges and complex three-phase, the brain biopsy post-morten showed severe loss of the neuronal population in all the layers, vacuoles in the neuropil, in the neuronal soma and the glial. The analysis of sequence of the gene PRNP identified homozygotes for methionine at codon 129. The patient died at 3 months of the onset of symptoms.
Conclusions: Epidemiology, clinical course and paraclinical examinations confirmed the diagnosis of Creutzfeldt-Jakob sporadic. The genotyping for polymorphisms of risk becomes useful tool to complement through molecular diagnosis and to deepen the understanding of the pathophysiology of Creutzfeldt- Jakob disease, both for sporadic forms and for the new variant.
Key words: Creutzfeldt-Jakob disease, prion protein, polymorphisms.
INTRODUCCIÓN
El presente estudio pretende entregar un análisis clínico y molecular, haciendo uso de diferentes metodologías diagnósticas, de una mujer diagnosticada con enfermedad de Creutzfeldt- Jakob esporádica.
Las encefalopatías espongiformes transmisibles (EET) o enfermedades priónicas son condiciones neurodegenerativas mortales que afectan tanto a seres humanos como a una amplia variedad de mamíferos (1-2). Aunque estas enfermedades se presentan con ciertas similitudes morfológicas y patológicas a las encontradas en la enfermedad de Alzheimer y la enfermedad de Parkinson, tienen como característica distintiva el ser transmitidas por inoculación o ingestión de material contaminado de una isoforma patogénica de la proteína priónica, la cual, en su estado normal, hace parte de los componentes de la membrana celular (3).
La forma mas común en humanos de las EET es la enfermedad Creutzfeldt-Jakob (ECJ), la cual ha sido clasificada como esporádica (esECJ), familiar (fECJ), iatrogénica (iECJ) y nueva variante (nvECJ) (4).
La forma esporádica es la más frecuente; representa un 90% de los casos y clásicamente ocurre generalmente entre los 50 y 70 años con un pico máximo de aparición a los 60. La forma familiar corresponde a un 10% y la iatrogénica, que aparece como producto de la acción contaminante de tejidos o material quirúrgico, representa menos del 1% (5).
ECJ esporádica
La esECJ es inusual, con una prevalencia de un caso por millón de habitantes, sin predominio estacional o geográfico; afecta tanto a hombres como mujeres por igual (1-6), aunque han sido reportadas incidencias más altas en algunos países como Suecia y Chile (7-8). La mayoría de los pacientes fallece al año de inicio de los síntomas neurológicos. El modo de infección es desconocido y su etiología no es muy clara ya que, hasta ahora, no han sido identificados agentes exógenos o endógenos que desencadenen la enfermedad (9). Muchos investigadores sugieren que la ECJ esporádica surge por generación endógena de priones, se supone que plegamientos anormales aleatorios de la proteína priónica podrían dar lugar a una serie de isoformas anormales, otros argumentan mutaciones somáticas en el gen que codifica para la proteína priónica PRNP (10).
ECJ familiar
Las formas familiares tienden a ocurrir a una edad más temprana que la esECJ, con una progresión más lenta. Son heredadas de manera autosómica dominante y cosegregan con mutaciones en el gen PRNP (11). Se han encontrado alrededor de 50 mutaciones en el gen PRNP en las familias con fECJ. Existen cuatro mutaciones puntuales ubicadas en los codones 102, 178, 200 y 210 y una inserción de octapéptidos repetidos que cubren el 95% de los casos (12). La mutación más común en las formas familiares es la que aparece en el codón 200; la clínica de las personas con esta mutación se asemeja al curso que siguen los pacientes con esECJ (13). Otras mutaciones generan fenotipos diferentes al del esECJ, para los cuales han sido utilizados otros nombres como Gerstmann-Sträussler-Scheinker e insomnio familiar fatal (14).
CJ iatrogénica
La iECJ es causada por la exposición de individuos a la proteína priónica durante procesos neuroquirúrgicos como el transplante de corneas o de duramadre y el tratamiento con extractos provenientes de pituitaria de cadáveres humanos. Es una entidad rara, con menos de 300 casos documentados, la mayor parte por implantación de duramadre o tratamiento con hormona de crecimiento de cadáver (15).
ECJ nueva variante
Es una nueva forma de encefalopatía espongiforme, reportada desde 1996. La mayoría de los casos han sido registrados en el Reino Unido, con unos pocos casos provenientes de otros países. Estudios moleculares y bioquímicos han determinado como agente causal de esta enfermedad al mismo que ocasiona los brotes de encefalopatía espongiforme bovina (BSE) en el ganado, también llamada enfermedad de las vacas locas (16-17).
Biología de la proteína prionica (PrPc)
La palabra prion se deriva del inglés proteinaceous infectious particle, que expresa el concepto de que este agente infeccioso consiste únicamente de una proteína carente de ácidos nucleicos. Se sabe que el agente infeccioso es más pequeño que la mayoría de los virus y es muy resistente al calor, los rayos ultravioleta y los desinfectantes comunes, los cuales, habitualmente, inactivan los virus (18).
La proteína priónica, en su versión normal (PrPc), después de ser sintetizada, se modifica en el aparato de Golgi y luego se transporta hacia la superficie celular (18); se encuentra presente en las membranas neuronales de los mamíferos. Estudios recientes han mostrado que está involucrada en la trasmisión sináptica, la transducción de señales, actividad antioxidante de la superoxidodismutasa, neuroplasticidad y sobrevida celular (19).
Para esta proteína se han identificado dos isoformas: la normal (PrPc) y la patológica o infecciosa (PrPsc). Con respecto al desarrollo de las encefalopatías priónicas, se sabe que un cambio en la estructura terciaria de la PrPc es la responsable del desarrollo de la enfermedad. La forma PrPSc sufre mayores plegamientos (tipo beta) con respecto la versión normal, PrPc, caracterizada por poseer plegamientos alfa y una minoría de estructuras beta (18). En su forma patógena, tiene la capacidad de generar un cambio en el plegamiento de la proteína normal, desencadenando una reacción en cadena de plegamientos anormales, haciendo, así, que se comporte como un agente infeccioso (20).
La isoforma normal ha sido identificada en tejidos de mamíferos, entre ellos: ovinos, bovinos, hamsters, ratón y en humanos, con un 80-90% de homología entre especies y del 98% en los genes de priones de ovino y bovino, lo cual explicaría el evento del paso de barrera inter-especies (21-22).
El gen codificante de la proteína priónica (PRNP) se encuentra en el brazo corto del cromosoma 20, región p12. Está compuesto por dos exones separados por un intrón de 12.696 pb. El exón I, constituido por 134 pb, es una región líder y no se traduce. El exón II, de 2355 pb, tiene un marco de lectura abierto, constituido por 759 nucleótidos que codifican para la proteína priónica completa. No existe, por tanto, la posibilidad de splicing o empalme alternativo, como tampoco generación de isoformas de PrP por esta vía (23-24). La región promotora del gen PRNP humano consta de un único punto de inicio de transcripción funcional y, por consiguiente, de una única proteína posible. (25).
Para este gen se han descrito 24 mutaciones puntuales, a partir del estudio del ADN aislado de pacientes con enfermedades priónicas y, por lo menos, 22 variantes asociados a una mayor susceptibilidad a la enfermedad, entre ellas 18 SNPs y 5 polimorfismos de longitud (25).
Diagnóstico clínico de las enfermedades priónicas en humanos
Dentro de las expresiones clínicas de la enfermedad de Creutzfeldt-Jakob existe un patrón común a todas: una encefalopatía espongiforme, la cual se asocia con la presencia en el tejido cerebral de una proteína malformada, que tiende a polimerizase y a depositarse como amiloide (8).
Al inicio de la enfermedad no existen síntomas específicos, lo cual genera dificultades diagnósticas; los más habituales son los cambios depresivos de la personalidad, trastornos del sueño y pérdida de peso. En la mayoría de los pacientes se observa deterioro de las funciones cognitivas y/o anomalías en la conducta.
En algunos pacientes la patología puede ir acompañada únicamente de déficit neurológico, con disfunción cerebelosa, en la cual la ataxia en la marcha es el signo más frecuente (26).
Cuando esta encefalopatía progresa, converge hacia un cuadro demencial subagudo, acompañado de deterioro visuocortical, en ocasiones, con diplopía o visión borrosa; además hay una disminución gradual de la memoria y una pérdida de las funciones cerebrales principales; también suelen aparecer mioclonías (contracciones musculares involuntarias sin alteración de la conciencia), que permiten que en un buen número de los casos se sospeche de ECJ, por primera vez. Los desencadenantes típicos de las mioclonías en estos pacientes son los ruidos fuertes y los estímulos táctiles (respuesta de sobresalto) (27).
Dentro del espectro de signos clínicos de la enfermedad, cuando ya está francamente desarrollada, se encuentran la disfunción piramidal (paresia espástica con signos del tracto piramidal) y extrapiramidal, y mutismo acinético, el cual se instaura en unas pocas semanas o meses; se caracteriza por ausencia de movimientos espontáneos, pérdida del habla e incapacidad de respuesta.
Finalmente se desarrolla una fase de disfunción generalizada, en la que todos los sistemas neurológicos fracasan y, tras un periodo de días en estado de coma, los pacientes fallecen (28).
Para hacer el diagnóstico de ECJ, además de la presentación clínica, se cuenta con:
1. Electroencefalograma en el cual se suele presentar una actividad especular seudoperiódica. Estos patrones, aunque son característicos, no son exclusivos de la ECJ y se presentan sólo durante un momento de la evolución por lo que su ausencia no excluye al diagnóstico (29).
2. Examen citoquímico del líquido cefalorraquídeo. En la mayoría de los casos suele ser normal. En 1996 se desarrolló una técnica para la identificación de una proteína denominada proteína priónica 14-3-3; sin embargo, los resultados son muy variables con una alto porcentaje de falsos positivos y negativos (30).
3. Resonancia magnética nuclear con técnicas de difusión que puede mostrar señales anormales de reforzamiento en áreas de la corteza cerebral y en los núcleos estriados (putamen y caudado). Estas regiones coinciden con los sitios de mayor alteración espongiforme por lo cual se consideran una buena guía para la biopsia cerebral diagnostica (31).
4. Estudio histopatológico del cerebro con el cual se puede establecer la encefalopatía espongiforme dada por la pérdida de neuronas, además de proliferación glial y placas amiloides (32). Por métodos de inmunohistoquímica también se puede identificar la presencia de la proteína priónica anormal en la muestra de biopsia.
CASO CLÍNICO
Se trata de una mujer de 58 años que consultó al servicio de psiquiatría por presentar un trastorno de conducta depresivo asociado con ansiedad y dolor inespecífico en miembros inferiores de 15 días de evolución. La sintomatología descrita empeoró con la aparición de alucinaciones auditivas por lo cual es hospitalizada. No se logró obtener un diagnóstico y la paciente firmó alta voluntaria. Un mes más tarde, es llevada por el esposo al servicio de urgencias por incremento de síntomas depresivos, mutismo, bradiquinesia, y bradipsiquia; se valoró nuevamente por el servicio de psiquiatría donde hicieron diagnóstico de síndrome mental orgánico e iniciaron tratamiento con fluoxetina, alprazolam y haloperidol. No hay mejoría y dos meses después aparece insomnio, vomito, llanto inmotivado, anorexia, mutismo y aquinesia.
Se realizaron estudios de imágenes diagnósticas (TAC cerebral simple), química sanguínea y orina, los cuales fueron reportados como normales; se continuó tratamiento psiquiátrico con haloperidol, fluotexina, alprazolam, biperideno, y risperidena; y se remitió a neurología con diagnóstico de síndrome mental orgánico.
En neurología, al examen físico, se encontró: alerta, con signos vitales estables, afebril, hidratada, mutista, aquinética, hace seguimiento visual, emite sonidos guturales, no repite, no nomina, se evidencia hipertonía generalizada con predominio en miembro superiores, reflejos osteotendinosos normales, presenta movimientos mioclónicos en reposo y al estimulo táctil.
Antecedentes de importancia: histerectomía hace 10 años por hemorragia uterina disfuncional, convive con loros hace 20 años, fue vacunada contra la fiebre amarilla hace 2 años.
Estudios paraclínicos de ingreso mostraron cuadro hemático y química sanguínea dentro de los límites normales; el líquido cefalorraquídeo con concentración de glucosa, proteínas y recuento celular dentro de parámetros normales también.
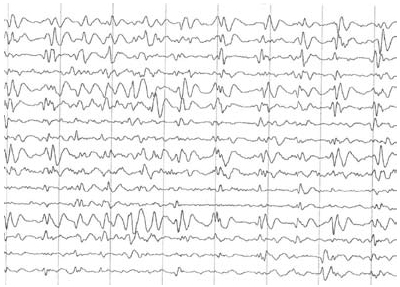
Se realizó resonancia magnética nuclear que mostró dilatación ventricular e híperintensidades inespecíficas córtico-subcorticales y en el núcleo estriado (figura 1). En el electroencefalograma de vigilia se evidenció pérdida de ritmos de fondo, patrón de descargas periódicas generalizadas y complejos trifásicos de 1 a 2 ciclos por segundo (figura 2).
Tres días después apareció ceguera cortical e hipertonía generalizada, por lo cual se decide hacer biopsia cerebral abierta, siguiendo protocolos de bioseguridad para enfermedad por priones.
Figura 1. Resonancia Nuclear Magnética de pobre calidad, hiperintensidades córtico-subcorticales y en el núcleo estriado de interpretación inespecífica. RMN tomada el día de ingreso a neurología
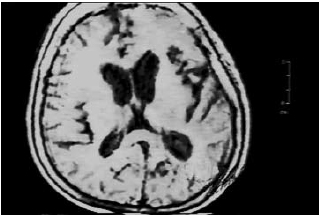
Figura 2. Electroencefalograma de vigilia. Muestra perdida de los ritmos de fondo, patrón de descargas generalizadas, que semejan complejos trifásicos de 1 a 2 ciclos por segundo.
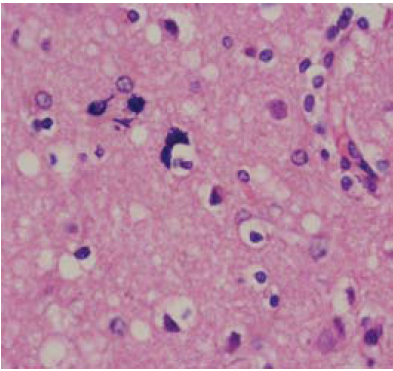
En el estudio histopatológico se encontró pérdida severa de la población neuronal en todas las capas, ausencia de inflamación, vacuolas de pequeño y gran tamaño, tanto en el neuropil, como en el soma neuronal y en la glía (figura 3). Los núcleos de los astrocitos se evidenciaron irregulares, hipertróficos, con procesos gruesos e irregulares (figura 4); no se observaron placas floridas de amiloide en el material examinado. Además, se hizo estudio de inmunoperoxidasas para la Prpsc humana que resulto negativo.
Figura 3. Biopsia cerebral, corteza cerebral con marcada pérdida neuronal, estado espongioso con vacuolas pequeñas a grandes pericelulares, ausencia de infiltrado inflamatorio (hematoxilina- eosina) X40
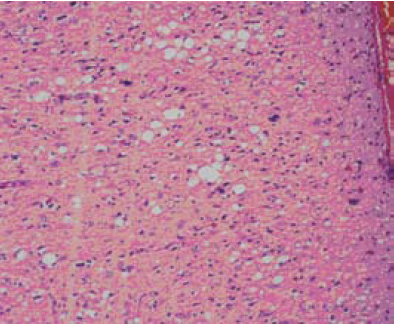
Figura 4. Biopsia cerebral, sustancia blanca con astrocitos de núcleos hipertróficos y procesos gruesos e irregulares (hematoxilina-eosina) x100.
Una semana después, se deterioró el estado de conciencia, variando entre estupor superficial y coma; además, aumentó el grado de disautomia, se presentaron movimientos clónicos y distónicos casi permanentes, presión arterial fluctuante; el coma se profundizó y finalmente la paciente falleció.
Estudio del gen PRNP
Para la determinación del genotipo del exón 2 se realizó extracción de DNA, amplificación y secuenciación de la paciente y de un voluntario sano para determinar el genotipo característico, comparándolo con la secuencia normal que reporta la base de datos del GENBANK.
Extracción del DNA
El DNA fue extraído a partir de 10 ml de sangre periférica, tomados de la paciente y un voluntario sano, previo consentimiento informado. La extracción del DNA se hizo utilizando el método PROBE (33).
Amplificación del DNA por PCR
Para la reacción en cadena de la polimerasa, se diseñaron un par de primers que permitieran amplificar la región genómica donde se concentra la mayoría de los polimorfismos reportados para el gen de la proteína priónica. Para este fin se utilizó el programa Web Primer versión libre (34).
La secuencia de los primers es: PR1-F 5´AAGCCTGGAGGATGGAACACTG3´ y PR1-R 5´ATGCTCGATCCTCTCTGGTAAT3´.
Éstos amplifican un fragmento de 617 pb, el cual corresponde a la mayor parte del éxon 2 (figura 5). Las condiciones de la reacción fueron: 94 C por 5 minutos, 35 ciclos de 94 C por 45 segundos, 62 C por 1 minuto, 68 C por 1 minuto y finalmente 5 minutos a 68C.
Secuenciación
Se hizo secuenciación de las dos hebras del producto amplificado con un kit de secuenciamiento directo ABI PRIMS (Applied Biosystems), que usa la enzima AmpliTaq DNA polimerasa. Las mezclas de la reacción de secuenciamiento fueron analizadas en un secuenciador automático (ABI PRIMS modelo 310), utilizando los primers y las condiciones reportadas previamente.
Figura 5. Principales poliformorfismos reportados para la proteína priónica. Se encuentran resaltados en su orden: Metionina como codón de inicio. Primer F. Polimorfismos prolina- leucina 102, alanina - valina 117, metionina - valina 129, aspártico - asparagina 178, fenilalanina - serina 198, glutamina - serina 200, valina - isoleucina 210, glutamina arginina 217. Primer R.

RESULTADOS Y DISCUSIÓN
El diagnóstico final, tanto por clínica como por histopatología de este caso, es ECJ esporádica. En el 90% de los pacientes, la es ECJ sigue un curso evolutivo subagudo, siendo fatal antes del año del inicio de los síntomas neurológicos; la mediana de sobrevida es de 4 meses; en este caso, la sobrevida fue alrededor de 3 meses.
Respecto a la clínica de inicio, ésta puede ser muy variada; en este caso, se presentó de manera inespecífica, con sintomatología general, acompañada de un trastorno depresivo, con alucinaciones auditivas e insomnio. Este tipo de presentación no es infrecuente, encontrándose que alrededor del 35% de los pacientes debutan con síntomas consistentes en fatiga, depresión, trastornos del sueño, pérdida de peso y apetito antes de la aparición del cuadro neurológico (35). Por lo anterior no es raro que estos pacientes sean estudiados por diferentes especialistas (psiquiatra, internista), antes de llegar al neurólogo.
La presentación clínica más frecuente es la de un deterioro cognitivo subagudo que se evidencia hasta en un 65% de los pacientes, en quienes se presenta compromiso de la memoria, trastornos cerebelosos y mioclonias. También existe una forma de presentación focal en la cual es posible identificar una alteración puntual del sistema nervioso. El más frecuente es el compromiso cerebeloso, seguido por la presentación con compromiso visual: el paciente se queja de disminución de la agudeza visual, alteración en la percepción de los colores o déficits campimétricos. El cuadro puede evolucionar a una ceguera cortical o síndrome de Balint (36). El caso reportado se presentó inicialmente con deterioro cognitivo y, casi al final del curso de la enfermedad, apareció una forma focalizada con ceguera de origen cortical.
Aunque no existe en la actualidad una prueba paraclínica que se pueda realizar en vida y que permita hacer un diagnóstico de certeza de esECJ, existen varios exámenes que, en el contexto clínico, son de gran valor diagnóstico: RMN en secuencias ponderadas en difusión, con la cual, recientemente, se han descrito anormalidades en la esECJ, principalmente un aumento en la señal de los ganglios basales y corticales (37). Las secuencias de difusión se basan en la modulación de la intensidad de señal causada por la difusión de las moléculas de agua, por lo tanto, los valores del coeficiente de difusión son la expresión de la alteración en la difusión del agua en el parénquima cerebral. Se cree que la alteración de la señal de difusión en los pacientes con ECJ corresponden con la pérdida neuronal, alteración de la citoarquitectura y cambios espongiformes (38).
Los cambios EEG pueden ser un apoyo diagnóstico importante, debido a que las alteraciones en pacientes con esECJ son progresivas, en etapas iniciales, existe un enlentecimiento del ritmo de base, posteriormente aparecen complejos periódicos bifásicos o trifásicos que, aunque son característicos, no son patognomónicos, pues se pueden presentar en otro tipo de demencias (39). En los periodos iniciales de la enfermedad, un 30% de los pacientes presentan los complejos periódicos; en estadios posteriores, éstos aparecen hasta en un 70% de los casos. En nuestro caso, se evidenciaron tanto las anormalidades en la RNM como los cambios EEG tardíos; estos últimos hallazgos, asociados a la clínica, especialmente las mioclonías y la ceguera cortical permitieron tener una alta certeza del diagnóstico de esECJ.
En la paciente no se realizó detección de proteína 14-3-3 en LCR, tanto por limitaciones de laboratorio como porque en el momento en que se tomó la muestra de LCR no había una sospecha clara de enfermedad por priones; después, con la evolución y el resultado de RMN y el EEG, no se consideró necesario repetir la punción lumbar.
En cuanto a la caracterización molecular del exón 2 del gen PRNP en la paciente y en el voluntario sano, se encontró homocigosis para todos los polimorfismos estudiados (figura 5), excepto para el 129 Met-Val, en el cual el voluntario sano resultó heterocigoto (figura 6). Lo anterior se hizo mediante la comparación de la secuencia obtenida con la reportada por el GENBANK (NM000311). Es de destacar que el polimorfismo 129, en el que hay un cambio ATG (Metionina) por CTG (Valina), influencia la expresión clínica de la enfermedad en diferentes aspectos como susceptibilidad, curso y patrones clínicos (35). La presencia de homocigotos Met-Met incrementa cuatro veces el riesgo de padecer la enfermedad y reduce a una tercera parte su duración, cuando hay heterocigotos. La prevalencia del homocigoto Met-Met en la población general es del 38%, la del heterocigoto Met-Valina es del 51% y la del homocigoto Val-Val es del 11%. Todos los casos reportados para nvECJ tienden a ser homocigotos Met-Met (40).
Figura 6. Comparación de las secuencias de la paciente (A) y el voluntario sano (B). La flecha señala el polimorfismo 129 Met/Val para el que la paciente es homocigota y el voluntario sano es heterocigoto.
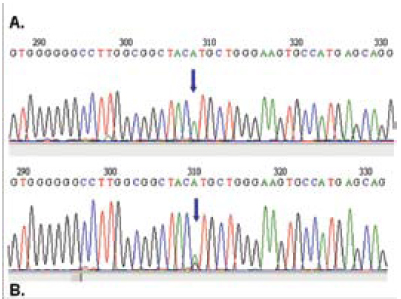
Aunque aún no está claro porque el alelo Met- Met es tan importante como marcador de riesgo para desarrollar ECJ esporádica o nueva variante, recientes investigaciones reportan interesantes avances en la comprensión, a nivel molecular, de los procesos involucrados en la patogénesis de la enfermedad en la personas Met-Met 129.
Estudios de oligomerización in vitro de las diferentes variantes de la proteína con respecto al codón 129 han determinado que la Met-Met 129 tiene una alta propensión a formar oligómeros ricos en hojas beta, a diferencia de la Val-Val 129, que tiene mayor tendencia a formar monómeros ricos en alfa hélices y que una mezcla equimolar de ambas variantes tiene un comportamiento de plegamiento intermedio con una inhibición de cerca del 30% de la formación de los oligomeros. Estas diferencias en el plegamiento para las diferentes variantes de la PRNP brindan una convincente explicación cinética a la alta susceptibilidad de individuos homocigotos para metionina (41); sin embargo, siguen quedando dudas acerca del significado biológico de estos hallazgos, pues se sabe que in vivo la proteína priónica mal plegada es resistente a la destrucción con proteinasas, contrario a lo que sucede con formas anómalas detectadas in vitro.
Lewis et al, en 2006, (42), buscando determinar precisamente las implicaciones biológicas directas de los diferentes alelos del polimorfismo, reportan que el alelo met 129 en una conformación parcialmente desnaturalizada (como podría ocurrir in vivo) tiene una profunda influencia en la habilidad de la proteína para formar fibrillas amiloides espontáneamente, generando, así, una clara conexión entre el polimorfismo 129 y el riesgo aumentado a desarrollar la enfermedad.
Por otra parte, por análisis filogenéticos, se ha identificado a la metionina como el aminoácido ancestral en el codón 129, y a la valina como una mutación característica del gen en humanos. Lo anterior asociado a la distribución de las frecuencias alélicas y a la diversidad de haplotipos en el locus PRNP indican una fuerte selección durante la evolución de los humanos modernos y que el estado de heterocigoto en el codón 129 confiere una ventaja selectiva significativa (43). Por esta razón, la genotipificación del gen PRPN no es sólo una poderosa herramienta para afinar diagnósticos individuales y para mejorar la compresión de la biología molecular de la enfermedad sino que también brinda información importante para determinar susceptibilidades poblacionales a ECJ, tanto esporádica como a la nueva variante.
REFERENCIAS
1. Aguzzi A, Montrasio F, Kaeser PS. Prions: health scare and biological challenge. Nat Rev Mol Cell Biol 2001; 2:118-26. [ Links ]
2. Aguzzi A, Polymenidou M. Mammalian prion biology: one century of evolving concepts. Cell 2004;116:313-27. [ Links ]
3. Aguzzi A, Haass C. Games played by rogue proteins in prion disorders and Alzheimers disease. Science 2003;302:814-18. [ Links ]
4. Glatzel M, Ott PM, Lindner T et al. Human prion diseases: epidemiology and integrated risk assessment. Lancet Neurol 2003;2:757-63. [ Links ]
5. Johnson RT, Gibbs CJ Jr. Creutzfeldt-Jakob diseaseand related transmissible spongiform encephalopathy. N Engl J Med 1998; 339:1994-04. [ Links ]
6. Brandel JP, Delasnerie-Laupretre N, Laplanche JL, Hauw JJ, Alperovitch A. Diagnosis of Creutzfeldt-Jakob disease: effect of clinical criteria on incidence estimates. Neurology 2000; 54:1095-99. [ Links ]
7. Glatzel M, Rogivue C, Ghani A, Streffer JR, Amsler L, Aguzzi A. Incidence of Creutzfeldt-Jakob disease in Switzerland. Lancet 2002; 360:139-41. [ Links ]
8. Cartier RL, Fernández OJ, Ramírez VE. Genetic markers in four Chilean families with familial Creutzfeldt- Jakob disease. Rev Méd Chile 2006;134: 1116-22. [ Links ]
9. Brown P, Cervenakova L, McShane L et al. Creutzfeldt-Jakob disease in a husband and wife. Neurology 1998; 50:684-88. [ Links ]
10. Johnson Richard T. Prion diseases. Lancet Neurol 2005; 4:635-42. [ Links ]
11. Hsiao K, Baker HF, Crow TJ et al. Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature 1989;338:342-45. [ Links ]
12. Capellari S, Cardone F, Notari S et al. Creutzfeldt-Jakob disease associated with the R208H mutation in the prion protein gene. Neurology 2005; 64: 905-07. [ Links ]
13. Mastrianni JA. Prion diseases and dementia. In: Power C, Johnson RT, eds. Emerging neurological infections. Boca Raton: Taylor and Francis, 2005, p. 77-13. [ Links ]
14. Masters CL, Gajdusek DC, Gibbs CJ Jr. Creutzfeldt-Jakob disease virus isolations from the Gerstmann- Sträussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virusinduced spongiform encephalopathies. Brain 1981; 104:559-88. [ Links ]
15. Brown P, Preece M, Brandel JP et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology 2000; 55:1075-81. [ Links ]
16. Aguzzi A. Between cows and monkeys. Nature 1996; 381:34. [ Links ]
17. Hill AF, Desbruslais M, Joiner S et al. The same prion strain causes vCJD and BSE. Nature 1997; 389:448- 50. [ Links ]
18. Prusiner SB. Prions. Proc Natl Acad Sci USA 1998; 95:13363-84. [ Links ]
19. Wang V, Chuang T, Hsu Y, Chou W, Kao M. Nitric oxide induces prion protein via MEK and p38 MAPK signalling. Biochem Biophys Res Commun 2005;333:95-00. [ Links ]
20. Prusiner SB, Scott MR. Genetics of prions. Annu Rev Genet 1997;31:139-75. [ Links ]
21. Premzl M, Sangiorgio L, Strumbo B, Marshall Graves JA, Simonic T, Gready JE. Shadoo, a new protein highly conserved from fish to mammals and with similarity to prion protein. Gene 2003;314C: 89-02. [ Links ]
22. Scott MR, Safar J, Telling G, Nguyen O, Groth D, Torchia M, Koehler R, Tremblay P, Walther D, Cohen FE, DeArmond SJ, Prusiner SB. Identification of a prion protein epitope modulating transmission of bovine spongiform encephalopathy prions to transgenic mice. Proc Natl Acad Sci 1997;94:14279-84. [ Links ]
23. Kretzsman HA, Stowring IE, Westaway D, Stubblebine WH, Prusiner SB. Molecular cloning of a human prion protein cDNA. DNA 1986;5:315-24. [ Links ]
24. Pucked C, Concannon P, Casey C. Genomic structure of the human prion protein gene. Am J Hum Genet 1991;49:320-29. [ Links ]
25. Soldevila M, Andres A, Ramírez-Soriano A, Márques-Bonet T, Calafell F, Navarro A, Bertranpetit J. The prion protein gene in humans revisited: Lesson from a worldwide resequencing study. Genome Res 2006;16:231-39. [ Links ]
26. Glatzel M, Stoeck K, Seeger H, Luhrs T, Aguzzi A. Human prion diseases: molecular and clinical aspects. Arch Neurol 2005;62:545-52. [ Links ]
27. Aguzzi A, Heikenwalder M, Miele G. Progress and problems in the biology, diagnosis, and therapeutics of prion diseases. J Clin Invest 2004;114:153-60. [ Links ]
28. Will RG, Mathews WB. A retrospective study of Creutzfeldt-Jakob disease in England and Wales 1970- 79, I: clinical features. J Neurol Neurosurg Psychiatry 1984;47:134-40. [ Links ]
29. Ortega-Albas JJ. Electroencefalograma en la enfermedad de Creutzfeldt-Jakob. Rev Neurol 2000;31:152-55. [ Links ]
30. Chapman T, McKeel DW, Morris IC. Misleading results with the 14-3-3 assay for the diagnosis of Creutzfeldt-Jakob disease. Neurology 2000;55:1396-98. [ Links ]
31. Demarel P, Sciot R, Robberecht W, Dorm R, Vandermeulen D, MAes F. Accuracy of diffusion-weighted MR imaging in the diagnostic of sporadic Creutzfeldt-Jakob disease. J Neurol 2003; 250:222-25. [ Links ]
32. Polo J. Historia y clasificación de las enfermedades criónicas humanas. Rev Neurol 2000;31:137-41. [ Links ]
33. Briceño I. Histocompatibility antigens and mitochondrial genome serological and molecular genetics studies of amerindian in Colombia. New Castle upon Tyne: New Castle, 1988. [ Links ]
34. Web Primer: DNA and purpose entry. En http://seq.yeastgenome.org/cgi-bin/web-primer. Acceso en: Oct, 25, 2008. [ Links ]
35. Moreno M, Romero J. La enfermedad de Creutzfeldt-Jakob esporádica: variabilidad fenotípica. Neurología 2002;17:336-77. [ Links ]
36. Brown P, Cathala F, Castaigne P, Gajdusek C. Creutzfeldt-Jakob disease: clinical analysis of a consecutive series of 230 neuropathologically verified cases. Ann Neurol 1986:20;597-02. [ Links ]
37. Rodríguez-Uranga J, Gil-Neciga E, Pinero P, Serrano-Pozo A. Diffusion-weighted magnetic resonance sequences in the early diagnosis of Creutzfeldt-Jakob disease. Rev Neurol 2005:41;16-30. [ Links ]
38. Moreno-Izco F, Martínez-Gil A. Creutzfeldt-Jakob disease: alterations in an isolated cortical signal in diffusion magnetic resonance imaging. Rev Neurol 2005:40;38-2. [ Links ]
39. Menéndez-González M, García-Fernández C, Suárez-San Martín E, Antón-González C, Blazquez-Menes B. Cronopatología de las enfermedades priónicas. Rev Neurol 2004; 39:962-5. [ Links ]
40. Dalsgaard N. Prion diseases. An overview. APMIS 2002;110:3-13. [ Links ]
41. Tahiri-Alaoui A, Gill A, Disterer P, James W. Methionine 129 variant of human prion proteína oligomerizes more rapidly than the valine 129 variant, implications for disease susceptibility to Creutzfeldt-Jakob disease. J Biol Chem 2004; 279:31390-97. [ Links ]
42. Lewis P, Tattum M, Jones S, Bhelt D, Bachelor M, Clarke A, Collinge J, Jackson G. Codon 129 polymorphism of the human prion proteína influences the kinetics of amyloid formation. J Gen Vir 2006; 87:2443-59. [ Links ]
43. Mead S, Stumpf M, Whitfield J, Beck J, Poulter M, Campbell T, Uphill J, Goldstein D, Alpers M, Fisher E, Collinge J. Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Sience 2003; 300:640-44. [ Links ]

