










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Ciencias de la Salud
versión impresa ISSN 1692-7273versión On-line ISSN 2145-4507
Rev. Cienc. Salud v.7 n.2 Bogotá mayo/ago. 2009
Papel de la vía fosfatidilinositol 3 kinasa (PI3K/Akt) en humanos
Role of phosphatidylinositol 3-kinase pathway (PI3K/Akt) in humans
Carlos Eduardo Pinzón, MD,1 Martha Lucía Serrano, PhD,2 María Carolina Sanabria, MSc3
1. Grupo de Genética Humana, Universidad del Rosario, Bogotá, Colombia. Grupo de Investigación Clínica, Instituto Nacional de Cancerología. Correo electrónico: cpinzon@cancer.gov.co, cepinzon@gmail.com.
2. Grupo de Investigación en Biología del Cáncer, Instituto Nacional de Cancerología, Bogotá, Colombia.
3. Ciencias Básicas Biomédicas. Universidad Industrial de Santander, Bucaramanga, Colombia.
Recibido: febrero 3 de 2009 Aceptado: junio 11 de 2009
Resumen
La vía de señalización de la fosfatidilinositol- 3-kinasa (PI3K) es crucial en numerosos aspectos del crecimiento y la supervivencia celular. Esta vía es estimulada fisiológicamente como consecuencia de muchos factores de crecimiento y factores reguladores. Varias alteraciones genéticas como amplificación, mutación y rearreglos cromosómicos pueden comprometer la vía PI3K, generando su activación permanente.
En diferentes tipos de cáncer se han encontrado evidencias de estas modificaciones genéticas deletéreas. La activación anormal de la vía PI3K resulta en alteración de los mecanismos de control del crecimiento y la supervivencia celular, lo que favorece el crecimiento competitivo, la capacidad metastasica y, frecuentemente, una mayor resistencia a los tratamientos. El objetivo de este articulo es revisar los aspectos relacionados con el funcionamiento de la vía de señalización PI3K/ Akt y su rol dentro del proceso de carcinogénesis en los seres humanos.
Palabras clave: Fosfatidilinositoles, Akt, PI3K, mTOR, vías de señalización, neoplasias.
Summary
The signaling pathway of phosphatidylinositol 3-kinase (PI3K) is critical in many aspects of growth and cell survival. The path of PI3K is stimulated physiologically as a result of many growth factors and regulatory factors. Several genetic alterations such as amplification, mutation and chromosomal arrangements may compromise the PI3K pathway, generating permanent activation in different cancer types have found evidence of these deleterious genetic modifications. Abnormal activation of the PI3K pathway results in alteration of the control mechanisms of growth and cell survival, which favors the competitive growth, and frequently metastatic capacity, greater resistance to treatment. The aim of this paper is to review matters relating to the operation of the PI3K/Akt signaling pathway and its role in the process of carcinogenesis in humans.
Key words: 1-Phosphatidylinositol 3-Kinase, Proto-Oncogene Proteins c-akt, PTEN Phosphohydrolase, m TOR protein, neoplasm.
INTRODUCCIÓN
La vía de señalización de la fosfatidilinositol- 3-kinasa (PI3K) es crucial en numerosos aspectos celulares involucrados en el crecimiento y la supervivencia celular (1). La vía de la PI3K es estimulada fisiológicamente como consecuencia de la activación de receptores de membrana tirosina kinasa, los cuales autofosforilan y fosforilan el sustrato del receptor de insulina (IRS); este ultimo, a la vez, fosforilara la subunidad p85 de la PI3K (Figura 1). La fosforilacion de la subunidad p85 conduce a un cambio conformacional de dicha proteína que conduce a la unión de la subunidad catalítica (p110). La PI3K activa, fosforila el fosfatidil inositol 3,4 difosfato (PIP2) convirtiéndolo en el segundo mensajero fosfatidil inositol 3,4,5 trifosfato (PIP3), el cual, corriente abajo, conduce a la activación de la proteína Akt. La Akt tiene múltiples blancos, responsables de los efectos de la activación de la vía (1-3). La activación anormal de esta vía conduce a una respuesta proliferativa y antiapoptotica que se relaciona con el desarrollo de múltiples tipos de cáncer; por esto, su estudio es parte crucial para el entendimiento de los procesos de carcinogénesis (4-5).
Figura 1. Esquema de la vía de señalización fosfatidil inositol 3 kinasa. ERK; kinasa regulada por señales extracelulares. FKHR; forkhead. GDP; ganosita difosfato. IRS; sustrato receptor de insulina. GSK3; kinasa sintasa de glicógeno. MAPK; protein kinasa mitógeno activado. NFkβ; factor nuclear kβ. PIP2; fosfatidil inositol 3,4 difosfato. PIP3; fosfatidil inositol 3,4,5 trifosfato. PKC; protein kinasa C. STATA; traductor de señal y activador de la trascripción Adaptado con permiso de Macmillan Publishers Ltd.: Exploiting the PI3K/Akt pathway for cancer drug discovery; Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB; Exploiting the PI3K/AKT pathway for cancer drug discovery, Nature reviews, drug discovery. Vol 4, 988-1004; 2005.
COMPONENTES DE LA VÍA P13K/AKT
PI3Ks
La familia de PI3K constituye un gran grupo de proteínas kinasas de serina/treonina, que incluyen fosfatidilinositol kinasas, proteínas kinasas dependientes de ADN (DNA-PK), ataxia telangiectasia-mutada (ATM) y ataxia telangiectasia y Rad3, relacionadas (ATR) (1,2,4). Existen tres tipos de PI3K. En la tabla 1 se enuncian las principales características de cada uno de los tipos de PI3K (6-21).
Tabla 1. Características de las isoformas de la proteína PI3K
AKT (proteína kinasa B)
Akt es el homologo humano del oncogén viral v-Akt (retrovirus Akt 8) y esta relacionado con proteínas kinasas A (PKA) y C (PKC) en humanos (22-23). Akt tiene tres isoformas conocidas, derivadas de distintos genes: Akt1/PKBα, Akt2/PKBβ y Akt3/PKBγ (23-24). El dominio PH en la región N-terminal de Akt interactúa con 3´-fosfoinositoles, contribuye al reclutamiento de Akt hacia la membrana plasmática (25). El dominio PH en la región N-terminal de Akt interactúa con 3´-fosfoinositoles, ayudando al reclutamiento de Akt hacia la membrana plasmática (24-26). Se han identificado diferentes PDK2 potenciales. Estas son proteinquinasas vinculadas en proceso de activación de diferentes vías de señalización, incluyendo el complejo rictor-mTOR (pero no el complejo raptor-mTOR inhibido por rapamicina y sus análogos), la ILK (kinasa ligada a integrina), la PKCβII e, incluso, la propia Akt, permitiendo, por tanto, un potente mecanismo de retroalimentación en la vía (27). Aun no hay claridad sobre el tipo de interacción de las PDK2 en la regulación de Akt; también falta esclarecer el papel relativo de la señalización de Akt en la membrana, el citosol y el núcleo (28-29).
Las isoformas de Akt se han visto implicadas en funciones especificas, relacionadas con cáncer (Akt2 en motilidad/invasión y Akt3 en la independencia hormonal) (30). En ratones knockout, la eliminación de las diferentes isoformas conlleva a diversos defectos en el desarrollo y alteraciones en la sensibilidad a la insulina (31- 32). Akt2 se encuentra amplificado en tumores pancreáticos, de mama y de ovario, y la actividad de Akt3 se ha visto aumentada, a través de un mecanismo desconocido, en canceres de próstata y de mama, no sensibles a hormonas (33).
Debido a que las aberraciones en Akt1 son mas frecuentes, Akt2 y Akt3 deben suplir diferentes funciones, generando, así, una presión selectiva para aberraciones durante la génesis tumoral. Los inhibidores selectivos de isoformas podrían ser necesarios para alcanzar una optima eficacia con una toxicidad aceptable (33).
Akt, los sensores de energía celular LKB1 (STK11) y la proteína kinasa AMP activada (AMPK) ejercen un efecto opositor en el blanco mamífero de la rapamicina (mTOR), activado por la Akt.
Eventos celulares corriente abajo, regulados por PI3K/Akt
La vía de PI3K se bifurca en muchos puntos, resultando en diversos resultados funcionales. La diversidad de respuestas funcionales probablemente representa los efectos del espectro, el nivel y la duración de la activación de componentes particulares de la vía de PI3K. Sin embargo, diferentes nodos en la vía se han implicado como elementos fundamentales en desenlaces particulares (Figura 1) (34).
Papel de PI3K en la supervivencia celular e inhibición de la apoptosis
Existen revisiones previas acerca del papel de la vía de PI3K/AKT en la proliferación y supervivencia celular (35-36). La señalización por Akt inactiva varios factores proapoptoticos como BAD, procaspasa-9 y factores de transcripción FKHR (Forkhead) (Figura 1) (37-38). Otros factores de transcripción que incrementan la expresión de genes anti-apoptoticos son activados por Akt, incluyendo CREB (proteína de unión a elementos de respuesta a AMP-cíclico), mediante fosforilacion directa, NF-KB e HIF-1α. La activación del factor de transcripción NF-KB, a través de Akt, se genera inicialmente mediante la fosforilacion y consecuente activa cion de IKKαβ (kinasa inhibidora de NF-KB), que fosforila y marca a IkB (inhibidor de NFkB) para ser degradado mediante el sistema ubiquitina-proteosoma, dejando libre a NF-KB, favoreciendo tanto su translocacion al núcleo, como su actividad de transcriptor de genes antiapoptoticos. Otro efecto de Akt, para favorecer la supervivencia celular, es la inactivación del gen supresor de tumor, p53; lo cual se da por la habilidad de Akt para fosforilar y activar directamente a MDM2, una proteína que regula negativamente a p53. MDM2 se encuentra usualmente en el citoplasma y tiene dominios NSL (señalización de localización nuclear) y de unión a p53. Cuando MDM2 es activado por Akt, se transloca al núcleo para unirse a p53, impidiendo su actividad como factor de transcripción de genes proapoptoticos; posteriormente, sale nuevamente al citoplasma donde media los procesos de ubiquitilacion y degradación proteosomica de p53, disminuyendo sus niveles. Cuando la vía PI3K/Akt se encuentra en un estado de activación permanente, el anterior mecanismo permite que una célula, aun en malas condiciones, resista a la apoptosis, sobreviva y prolifere, contribuyendo, de esta manera, a la inestabilidad cromosómica, característica de la vía supresora de la carcinogénesis (39-42). La fosforilacion de muchos efectos por Akt regula su localización y, por tanto, su actividad, al generar sitios de ligando para proteínas 14-3-3, importantes en la regulación de la localización celular y en la degradación de diversas moléculas (42-43).
Papel de PI3K en la transcripción de genes, traducción a proteínas, metabolismo, crecimiento y proliferación celular
La proteína serina/treonina kinasa, mTOR (blanco mamífero de rapamicina) activada por Akt, se puede considerar como el regulador central del crecimiento celular (44). mTOR regula la traducción proteica, en respuesta a nutrientes y factores de crecimiento, al fosforilar componentes de la maquinaria de la síntesis de proteínas como son p70S6K (proteína ribosomal S6 kinasa) y 4E-BP (proteína ligadora de 4E). La fosforilacion de 4E-BP1 por raptor-mTOR (proteína regulatoria asociada a mTOR), libera el eIF-4E (factor iniciador de la traducción eucariotico 4E) para restablecer la traducción a proteínas dependiente de cap. Se han evidenciado, además, las actividades transformadoras y anti-apoptoticas in-vitro de eIF-4E (45-47). El complejo formado por TSC1 (hamartina) y TSC2 (tiberina) es un heterodimero que regula negativamente la vía raptor/ mTOR, ya que, estando activo, retiene e inactiva la proteína Rheb. Estando activa, dicha proteína se une al complejo raptor/mTOR, activándolo y favoreciendo los procesos de traducción proteica y crecimiento celular. El mecanismo anterior sucede gracias a la actividad kinasa de Akt, que fosforila la subunidad TSC2 del complejo, inactivándolo y dejando libre y activa a la proteína Rheb para unirse el complejo raptor/mTOR (48- 50). La cascada TSC/Rheb/mTOR/S6K también regula el sustrato del receptor de insulina (IRS). y PDGFR, el cual incluye potencialmente una importante asa de retroalimentación adicional (51-52). De hecho, la inhibición de mTOR por rapamicina, o sus análogos, puede activar proteínas upstream, incluyendo Akt, lo cual, probablemente, es resultado de la perdida de la inhibición de la retroalimentación (53-54). La rapamicina y sus análogos inhiben la vía raptor/mTOR, es decir, bloquean la fosforilacion de p70S6K y de 4E-BP1. De esta manera, se impide la activación de p70S6K y la inhibición de 4E-BP1, regulando de forma negativa los procesos de traducción proteica y crecimiento celular, siendo este el efecto deseado sobre un tejido tumoral (55).
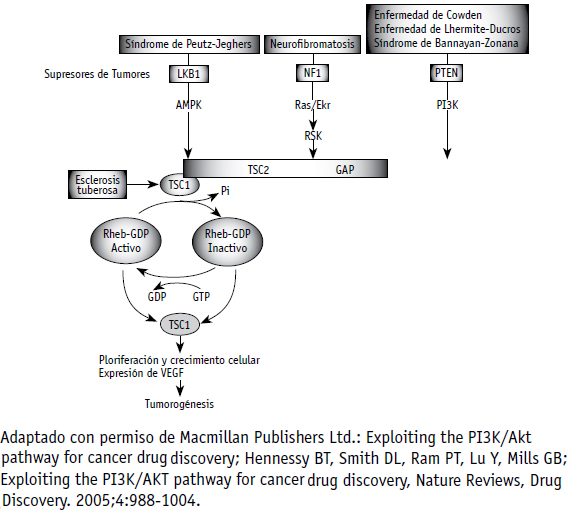
Adicionalmente, la vía de PI3K/Akt interactúa con los mecanismos de control de la energía celular y el metabolismo de la glucosa (Figura 2) (56). Cuando se genera estrés celular, por depleción de glucosa y/o hipoxia, aumentan los niveles de AMP y disminuyen los de ATP por falta en la producción de energía; es decir, hay una alteración en la relación AMP/ATP. La proteína LKB1 (STK11, kinasa serina treonina 11) forma un complejo con STRAD (proteína adaptadora relacionada a Ste-20) y MO25 (proteína murina 25), LKB1-STRAD-MO25, forma mediante la cual se hace mas activa. Este complejo fosforila a AMPK (proteína kinasa activada por AMP), sensor de estrés celular, y lo activa. AMPK ejerce, entonces, su actividad kinasa sobre TSC2, activando el complejo TSC1/TSC2, lo que permite el secuestro de Rheb, el consecuente bloqueo de la vía raptor/mTOR y, por tanto, del crecimiento celular, permitiendo, así, la conservación energética en condiciones adversas (Figura 3) (57). Mutaciones en la línea germinal de genes que codifican para PTEN (homologo de la fosfatasa y tensina), TSC2 y LKB, resultan en síndromes clínicos similares, caracterizados por la presencia de hamartomas en diferentes sitios, además de un incremento en el riesgo de aparición de canceres específicos. De esta manera, otorgan evidencia sobre la perdida de la regulación del crecimiento celular ante la desregulación de la vía de PI3K/ AKT, causada por la inactivación de estos genes cruciales (Figura 3) (58). Además, Akt esta involucrado en los procesos de regulación del ciclo celular al fosforilar las CKI (inhibidores de kinasas dependientes de ciclinas) p21CIP1/WAF1 y p27KIP1, favoreciendo, de esta manera, su translocacion al citoplasma y posterior degradación (59-61). Como resultado, se incrementa la proliferación celular, al abolirse el efecto antiproliferativo de las CKI sobre los complejos de ciclina-kinasas, y también, probablemente, a funciones citosolicas novedosas de los CKI. Akt, de forma directa o indirecta, fosforila e inhibe la GSK3 (kinasa glucógeno sintetasa-3), la fosfodiesterasa-3B, la proteína fosfatasa 2A y, posiblemente, a Raf1; en consecuencia, crea una compleja red intracelular (62). Los inhibidores de GSK3 previenen la fosforilacion de β-catenin, impidiendo su degradación, resultando en traslocacion al núcleo para estimular la trascripción de genes blanco, incluyendo c-JUN y homebox del gen CDX1 (63). Adicionalmente, la fosforilacion de ciclina D1 por GSK3 resulta en su desestabilización (64- 65). La señalización de PI3K también controla la angiogenesis, el crecimiento, la proliferación, la senescencia y otros procesos, a través de mecanismos como la activación transcripcional del factor de crecimiento vascular endotelial (VEGF) y la expresión del factor 1α inductor de hipoxia (HIF1 á) (66-67).
Regulación negativa de la vía PI3/Akt mediada por PTEN
PTEN es una proteína que regula la señal de supervivencia celular, dependiente e independiente de la vía PI3K/Akt. Esta proteína supresora de tumor se expresa cuando existen señalizaciones de daño celular, bloqueando la supervivencia de la célula mediante p53. La actividad fosfatasa de PTEN le permite desfosforilar la PI(3)P, inhibiendo el proceso de activación de Akt y el proceso de inactivación de p53 vía Akt-MDM2. PTEN también se une a p53, favoreciendo su estabilidad e impidiendo la actividad inhibitoria de MDM2 sobre la misma. Lo anterior representa que p53 se mantiene activa para inhibir la proliferación celular y promover los procesos de reparación o apoptosis. De esta manera, se protege tanto la integridad genómica como cromosómica de una célula y un tejido en general (68).
Figura 2. El rol de AKT y el blanco mamífero de la rapamicina (mTOR) en la homeostasis de la glucosa
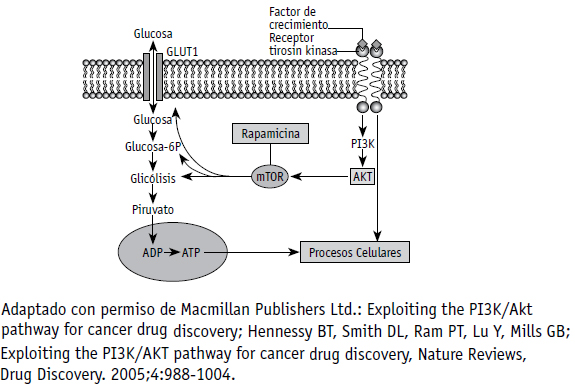
Figura 3. Síndromes clínicos asociados con la alteración de la vía de señalización PI3K/Akt, resultado se las mutaciones en cuatro genes supresores de tumor
Papel de la vía PI3K/Akt en cáncer
La inhibición de los componentes de la vía PI3K puede hacer oposición o sobreponerse a la resistencia, a quimioterapia, radioterapia, terapia hormonal y a los agentes dirigidos en cáncer (69-71). Un mecanismo potencial para esta oposición es la potenciación de la apoptosis. Sin embargo, la mayoría de estos estudios se han realizado con compuestos relativamente no específicos y podrían reflejar la inhibición de kinasas similares a PI3K, activadas por daño o irradiación al ADN (por ejemplo, proteína kinasa activada por ADN (DNA-PK), ATM y ATR), por medio de medicamentos tales como LY294002 o wortmannin (72-74).
En cáncer de seno existen importantes interacciones entre los efectos genómicos y no genómicos del receptor de estrógenos y las kinasas de membrana y citoplasmáticas, incluyendo a los miembros de la vía PI3K que juegan un papel en la resistencia anti-estrogenica (75-76). La actividad de PTEN contribuye a la eficacia de trastuzumab (Herceptin; Genetech) y a la radioterapia (77-78). Por tanto, el estado de activación de la vía de PI3K contribuye a la resistencia tumoral ante ciertas terapias, lo mismo que ante quimio y radioterapia (79).
Aberraciones y alteraciones genéticas de la vía de PI3K en cáncer
Las anomalías en la vía de PI3K son comunes en cáncer y participan en la transformación neoplasica (Tabla 2) (80). En si, PI3K es un blanco frecuente de activación mutacional (81). Las mas frecuentes aberraciones genéticas en cáncer de seno son las mutaciones missense somáticas en el gen que codifica p110a (PIK3CA). Estas mutaciones ocurren con mayor frecuencia en canceres HER2-amplificados y en positivos para receptor hormonal (82). La amplificación o mutación del gen PIK3CA también ocurren comúnmente en cáncer de colon, ovario (mutaciones en subtipos de células endometriales y claras, y amplificación en tumores serosos), de cabeza y cuello y escamoso de cuello uterino, gástrico, pulmonar, oligodendroglioma anaplasico, astrocitoma anaplasico, glioblastoma multiforme y meduloblastoma (82-87). A pesar de que las mutaciones y translocaciones en p85α son raras, sirven para hacer énfasis en la importancia de la vía (88).
La Akt y PTEN también son blanco de frecuentes cambios genómicos y epigeneticos en canceres humanos (Tabla 3) (89-99). Recientemente, y de forma sistemática, se realizo una secuenciación genética a gran escala de las fosfatasas y tirosin kinasas por parte de tres grupos académicos (ver el Sanger Centre Catalogue of Somatic Mutations in Cancer) (100-103). Se detectaron frecuentes mutaciones no descritas en la vía de PI3K o en otras vías de kinasas, por ejemplo, en los tres genes que codifican los componentes de la vía PI3K/Akt (PDK1, AKT2 y kinasa activada por p21 (PAK4)) en cáncer colorrectal. En modelos específicos, se demostró que las isoformas de Akt tienen actividad de transformación. Las alteraciones genéticas también se presentan en moléculas de otras vías relacionadas con cáncer (Tabla 4) (104-118). En muchos casos, los efectos oncogénicos de estas anormalidades están mediados, al menos parcialmente, por la señalización de PI3K/Akt. Mutaciones en Ras, que activan PI3K, son comunes en cáncer de páncreas (104-107).
La modulación de la vía PI3K/Akt es requerida para la génesis tumoral mediada por HER2, en modelos animales y para las respuestas con trastuzumab (109-113). La vía PI3K/Akt también se requiere para los efectos oncogénicos de EGFR. Un incremento en la actividad de PI3K esta asociado con transformación por SRC, antígeno T medio de polioma, ABl y Ros.
Tabla 2. Anormalidades en la vía de señalización PI3K/Akt en cáncer
Tabla 3. Anormalidades en las vías de señalización relacionadas con PI3K/Akt en cáncer
Tabla 4. Funciones asociados a las diferentes isoformas de p110 de PI3K
Importancia del estudio de la vía de Señalización
En contraste con p53 y otras vías supresoras tumorales, la vía del PI3K se activa en el cáncer, convirtiéndola en un blanco terapéutico optimo, ya que es mas fácil inhibir los eventos de activación que reemplazar la perdida de la función supresora de tumores (118). Mas de 20 compañías farmacéuticas y centros académicos han manifestado programas activos en esta área y se espera que en los próximos anos inicien ensayos clínicos de medicamentos específicos.
Algunos fármacos de uso clínico, o en fase de evaluación preclínica, originalmente desarrollados con otros propósitos o no identificados en los tamizajes de la vía PI3K, han demostrado, directa o indirectamente, actuar sobre la señalización de la vía (118-119). Estos incluyen inhibidores del blanco de la rapamicina mamífera (mTOR) de la familia rapalog, de los análogos de la rapamicina, los lípidos éter (tales como perifosina y miltefosina), y los inhibidores del receptor del factor de crecimiento epidérmico (EGFR), del HER2/ neu, del c-Kit, del receptor del factor de crecimiento derivado de plaquetas (PDGFR) y del BCR-ABL (119). Sin embargo, a excepción de los inhibidores del mTOR, que parecen actuar únicamente sobre la vía del PI3K, se desconoce si los desenlaces funcionales de estos medicamentos están relacionados con una inhibición de la vía del PI3K o con otros efectos. Dado que la vía del PI3K es importante para numerosas funciones celulares normales y, en particular, para la vía de señalización de la insulina, la mayor limitante para la implementación de medicamentos que inhiban esta vía, probablemente, será la identificación de blancos y fármacos que garanticen un índice terapéutico suficiente y confiable en la practica clínica (119).
Se desconoce si estos medicamentos podrán demostrar una actividad anti-tumoral de forma independiente o en combinación con otros agentes (120). En particular, debido a que la vía PI3K es un regulador crucial para la supervivencia durante estrés celular –y teniendo en cuenta que los tumores frecuentemente existen en ambientes intrínsecamente estresantes, con limitaciones en el suministro de oxigeno y de nutrientes, al igual que bajo pH– la inhibición de la vía PI3K probablemente pueda hallar una eficacia optima en combinación, con el fin de inducir estrés celular, incluyendo combinación con otros inhibidores de la señal de transducción y con quimioterapia o irradiación (119).
A medida que los inhibidores de la vía de PI3K sean integrados en la práctica clínica, será crucial desarrollar métodos para identificar a aquellos pacientes que se puedan beneficiar de estas terapias, desarrollando, paralelamente, marcadores moleculares y terapéuticas dirigidas (120). Esta aproximación maximizara la eficacia y costo-efectividad de estas nuevas terapias, y minimizara una innecesaria exposición de los pacientes (120). Esta aproximación fue muy exitosa con imatinib mesylate (Gleevec; Novartis), desarrollado para el tratamiento de leucemia mieloide crónica (CML), basado en que la CML es casi exclusivamente definida por la fusión del oncogén BCR-ABL. Posteriormente se encontró que el imatinib era altamente efectivo cuando actuaba sobre aberraciones genéticas en otras enfermedades tales como la activación mutacional de c-Kit en los tumores estromales gastrointestinales (GIST) y el PDGFR en dermatosarcoma protuberans y en el síndrome hipereosinofilico (120).
CONCLUSIONES
En las dos últimas décadas, la vía de señalización PI3K/Akt se ha establecido como un contribuidor critico en el proceso de tumorogenesis. La elucidación del rol de la vía de señalización en el crecimiento celular, la supervivencia y la proliferación ha abierto una puerta en el estudio de su regulación en el proceso tumoral. Componentes de la vía de señalización PI3K/Akt tienen compromisos emergentes como: nuevos blancos en el desarrollo de terapias en cáncer y la generación de inhibidores de la vía han mostrado ser efectivos, in vitro e in vivo, en la reducción del crecimiento tumoral. Se deberá continuar con esfuerzos para el desarrollo de inhibidores específicos, y con alta afinidad, para la vía PI3K/Akt, para generar un tratamiento efectivo para el cáncer en los seres humanos.
AGRADECIMIENTOS
Agradecemos al Instituto Nacional de Cancerología por su colaboración en el desarrollo de este articulo.
CONTRIBUCIONES DE LOS AUTORES
CP y MCS realizaron la búsqueda y lectura revisada para el manuscrito. CP realizo el primer manuscrito. CP realizo los procedimientos para la solicitud del permiso al autor para la modificación y el uso de su material publicado, y genero las adaptaciones de las figuras y tablas del manuscrito. MLS contribuyo en la designación de los estudios y el concepto de cada uno de ellos. Todos los autores leyeron y aprobaron el manuscrito final.
CONFLICTOS DE INTERÉS
No declarados.
REFERENCIAS
1. Hiles ID, Otsu M, Volinia S, Fry MJ, Gout I, Dhand R, et al. Phosphatidylinositol 3-kinase:structure and expression of the 110 kd catalytic subunit. Cell. 1992;70(3):419-29. [ Links ]
2. Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481-507. [ Links ]
3. Jimenez C, Jones DR, Rodriguez-Viciana P, Gonzalez-Garcia A, Leonardo E, Wennstrom S, et al. Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3-kinase. Embo J. 1998;17(3):743-53. [ Links ]
4. Oudit GY, Sun H, Kerfant BG, Crackower MA, Penninger JM, Backx PH. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J Mol Cell Cardiol. 2004;37(2):449-71. [ Links ]
5. Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000; 351:95-105. [ Links ]
6. Pawson T, Nash P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000 May 1;14(9):1027-47. [ Links ]
7. Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J Biol Chem. 1999 Apr 16; 274(16):10963-8. [ Links ]
8. Brachmann SM, Ueki K, Engelman JA, Kahn RC, Cantley LC. Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol Cell Biol. 2005 Mar; 25(5):1596-607. [ Links ]
9. Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002 Aug 9;297(5583):1031-4. [ Links ]
10. Bilancio A, Okkenhaug K, Camps M, Emery JL, Ruckle T, Rommel C, et al. Key role of the p110delta isoform of PI3K in B-cell antigen and IL-4 receptor signaling:comparative analysis of genetic and pharmacologic interference with p110delta function in B cells. Blood. 2006 Jan 15;107(2):642-50. [ Links ]
11. Northcott CA, Hayflick J, Watts SW. Upregulated function of phosphatidylinositol-3-kinase in genetically hypertensive rats:a moderator of arterial hypercontractility. Clin Exp Pharmacol Physiol. 2005 Oct;32(10):851-8. [ Links ]
12. Condliffe AM, Davidson K, Anderson KE, Ellson CD, Crabbe T, Okkenhaug K, et al. Sequential activation of class IB and class IA PI3K is important for the primed respiratory burst of human but not murine neutrophils. Blood. 2005 Aug 15;106(4):1432-40. [ Links ]
13. Sujobert P, Bardet V, Cornillet-Lefebvre P, Hayflick JS, Prie N, Verdier F, et al. Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood. 2005 Aug 1;106(3):1063-6. [ Links ]
14. Geng L, Tan J, Himmelfarb E, Schueneman A, Niermann K, Brousal J, et al. A specific antagonist of the p110delta catalytic component of phosphatidylinositol 3-kinase, IC486068, enhances radiation-induced tumor vascular destruction. Cancer Res. 2004 Jul 15;64(14):4893-9. [ Links ]
15. Yip SC, El-Sibai M, Hill KM, Wu H, Fu Z, Condeelis JS, et al. Over-expression of the p110beta but not p110alpha isoform of PI 3-kinase inhibits motility in breast cancer cells. Cell Motil Cytoskeleton. 2004 Nov;59(3):180-8. [ Links ]
16. Chantry D, Vojtek A, Kashishian A, Holtzman DA, Wood C, Gray PW, et al. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem. 1997 Aug 1;272(31):19236-41. [ Links ]
17. Gukovsky I, Cheng JH, Nam KJ, Lee OT, Lugea A, Fischer L, et al. Phosphatidylinositide 3-kinase gamma regulates key pathologic responses to cholecystokinin in pancreatic acinar cells. Gastroenterology. 2004 Feb;126(2):554-66. [ Links ]
18. Leverrier Y, Okkenhaug K, Sawyer C, Bilancio A, Vanhaesebroeck B, Ridley AJ. Class I phosphoinositide 3-kinase p110beta is required for apoptotic cell and Fcgamma receptor-mediated phagocytosis by macrophages. J Biol Chem. 2003 Oct 3;278(40):38437-42. [ Links ]
19. Campbell M, Allen WE, Sawyer C, Vanhaesebroeck B, Trimble ER. Glucose-potentiated chemotaxis in human vascular smooth muscle is dependent on cross-talk between the PI3K and MAPK signaling pathways. Circ Res. 2004 Aug 20;95(4):380-8. [ Links ]
20. Perrino C, Naga Prasad SV, Patel M, Wolf MJ, Rockman HA. Targeted inhibition of beta-adrenergic receptor kinase-1-associated phosphoinositide-3 kinase activity preserves beta-adrenergic receptor signaling and prolongs survival in heart failure induced by calsequestrin overexpression. J Am Coll Cardiol. 2005 Jun 7;45(11):1862-70. [ Links ]
21. Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, et al. PI 3-kinase p110beta:a new target for antithrombotic therapy. Nat Med. 2005 May;11(5):507-14. [ Links ]
22. Coffer PJ, Woodgett JR. Molecular cloning and characterisation of a novel putative protein-serine kinase related to the cAMP-dependent and protein kinase C families. Eur J Biochem. 1991 Oct 15;201(2):475-81. [ Links ]
23. Murthy SS, Tosolini A, Taguchi T, Testa JR. Mapping of AKT3, encoding a member of the Akt/protein kinase B family, to human and rodent chromosomes by fluorescence in situ hybridization. Cytogenet Cell Genet. 2000;88(1-2):38-40. [ Links ]
24. Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, et al. Mechanism of activation of protein kinase B by insulin and IGF-1. Embo J. 1996 Dec 2;15(23):6541-51. [ Links ]
25. Bellacosa A, Testa JR, Staal SP, Tsichlis PN. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991 Oct 11;254(5029):274-7. [ Links ]
26. Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001 May 17;411(6835):355-65. [ Links ]
27. Lynch DK, Ellis CA, Edwards PA, Hiles ID. Integrin-linked kinase regulates phosphorylation of serine 473 of protein kinase B by an indirect mechanism. Oncogene. 1999 Dec 23;18(56):8024-32. [ Links ]
28. Kawakami Y, Nishimoto H, Kitaura J, Maeda-Yamamoto M, Kato RM, Littman DR, et al. Protein kinase C betaII regulates Akt phosphorylation on Ser-473 in a cell type- and stimulus-specific fashion. J Biol Chem. 2004 Nov 12;279(46):47720-5. [ Links ]
29. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005 Feb 18;307(5712):1098-101. [ Links ]
30. Arboleda MJ, Lyons JF, Kabbinavar FF, Bray MR, Snow BE, Ayala R, et al. Overexpression of AKT2/ protein kinase Bbeta leads to up-regulation of beta1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003 Jan 1;63(1):196-206. [ Links ]
31. Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci USA. 2001 Sep 25;98(20):10983-5. [ Links ]
32. Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001 Oct 19;276(42):38349-52. [ Links ]
33. Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, Jacobs J, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003 Jun 1;17(11):1352-65. [ Links ]
34. Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E, et al. Protein kinase B alpha/ Akt1 regulates placental development and fetal growth. J Biol Chem. 2003 Aug 22;278(34):32124-31. [ Links ]
35. Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002 Jul;2(7):489-501. [ Links ]
36. Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer:rationale and promise. Cancer Cell. 2003 Oct;4(4):257-62. [ Links ]
37. Chen X, Thakkar H, Tyan F, Gim S, Robinson H, Lee C, et al. Constitutively active Akt is an important regulator of TRAIL sensitivity in prostate cancer. Oncogene. 2001 Sep 20;20(42):6073-83. [ Links ]
38. Wang Q, Wang X, Hernandez A, Hellmich MR, Gatalica Z, Evers BM. Regulation of TRAIL expression by the phosphatidylinositol 3-kinase/Akt/GSK-3 pathway in human colon cancer cells. J Biol Chem. 2002 Sep 27;277(39):36602-10. [ Links ]
39. Carroll PE, Okuda M, Horn HF, Biddinger P, Stambrook PJ, Gleich LL, et al. Centrosome hyperamplification in human cancer:chromosome instability induced by p53 mutation and/or Mdm2 overexpression. Oncogene. 1999 Mar 18;18(11):1935-44. [ Links ]
40. Toi M, Saji S, Suzuki A, Yamamoto Y, Tominaga T. MDM2 in Breast Cancer. Breast Cancer. 1997 Dec 25;4(4):264-8. [ Links ]
41. Sherr CJ, Weber JD. The ARF/p53 pathway. Curr Opin Genet Dev. 2000 Feb;10(1):94-9. [ Links ]
42. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999 Mar 19;96(6):857-68. [ Links ]
43. Cahill CM, Tzivion G, Nasrin N, Ogg S, Dore J, Ruvkun G, et al. Phosphatidylinositol 3-kinase signaling inhibits DAF-16 DNA binding and function via 14-3-3-dependent and 14-3-3-independent pathways. J Biol Chem. 2001 Apr 20;276(16):13402-10. [ Links ]
44. Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004 Mar 18;428(6980):332-7. [ Links ]
45. Schmelzle T, Hall MN. TOR, a central controller of cell growth. Cell. 2000 Oct 13;103(2):253-62. [ Links ]
46. Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5 cap. Nature. 1990 Jun 7;345(6275):544-7. [ Links ]
47. Polunovsky VA, Gingras AC, Sonenberg N, Peterson M, Tan A, Rubins JB, et al. Translational control of the antiapoptotic function of Ras. J Biol Chem. 2000 Aug 11;275(32):24776-80. [ Links ]
48. Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol. 2002 Sep;4(9):658-65. [ Links ]
49. Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci USA. 2002 Oct 15;99(21):13571-6. [ Links ]
50. Dan HC, Sun M, Yang L, Feldman RI, Sui XM, Ou CC, et al. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J Biol Chem. 2002 Sep 20;277(38):35364-70. [ Links ]
51. Hengstschlager M, Rosner M, Fountoulakis M, Lubec G. Tuberous sclerosis genes regulate cellular 14-3-3 protein levels. Biochem Biophys Res Commun. 2003 Dec 19;312(3):676-83. [ Links ]
52. Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004 Jul 19;166(2):213-23. [ Links ]
53. Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, et al. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest. 2003 Oct;112(8):1223-33. [ Links ]
54. Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005 Aug 15;65(16):7052-8. [ Links ]
55. Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005 Sep;8(3):179-83. [ Links ]
56. Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003 Jul 3;546(1):113-20. [ Links ]
57. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004 Mar 9;101(10):3329-35. [ Links ]
58. Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, et al. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004 Jul;6(1):91-9. [ Links ]
59. Fujita N, Sato S, Tsuruo T. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2003 Dec 5;278(49):49254-60. [ Links ]
60. Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med. 2002 Oct;8(10):1145-52. [ Links ]
61. Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Aktinduced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001 Mar;3(3):245-52. [ Links ]
62. Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998 Nov 15;12(22):3499-511. [ Links ]
63. Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004 Dec 15;22(24):4991- 5004. [ Links ]
64. Skinner HD, Zheng JZ, Fang J, Agani F, Jiang BH. Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J Biol Chem. 2004 Oct 29;279(44):45643-51. [ Links ]
65. Zhang D, Brodt P. Type 1 insulin-like growth factor regulates MT1-MMP synthesis and tumor invasion via PI 3-kinase/Akt signaling. Oncogene. 2003 Feb 20;22(7):974-82. [ Links ]
66. Kimura A, Ohmichi M, Kawagoe J, Kyo S, Mabuchi S, Takahashi T, et al. Induction of hTERT expression and phosphorylation by estrogen via Akt cascade in human ovarian cancer cell lines. Oncogene. 2004 Jun 3;23(26):4505-15. [ Links ]
67. Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat Cell Biol. 2004 Apr;6(4):358-65. [ Links ]
68. Zhou M, Gu L, Findley HW, Jiang R, Woods WG. PTEN reverses MDM2-mediated chemotherapy resistance by interacting with p53 in acute lymphoblastic leukemia cells. Cancer Res. 2003 Oct 1;63(19):6357-62. [ Links ]
69. Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, et al. Mechanisms of tamoxifen resistance:increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004 Jun 16;96(12):926-35. [ Links ]
70. Mondesire WH, Jian W, Zhang H, Ensor J, Hung MC, Mills GB, et al. Targeting mammalian target of rapamycin synergistically enhances chemotherapy-induced cytotoxicity in breast cancer cells. Clin Cancer Res. 2004 Oct 15;10(20):7031-42. [ Links ]
71. Knuefermann C, Lu Y, Liu B, Jin W, Liang K, Wu L, et al. HER2/PI-3K/Akt activation leads to a multidrug resistance in human breast adenocarcinoma cells. Oncogene. 2003 May 22;22(21):3205-12. [ Links ]
72. Gupta AK, Cerniglia GJ, Mick R, Ahmed MS, Bakanauskas VJ, Muschel RJ, et al. Radiation sensitization of human cancer cells in vivo by inhibiting the activity of PI3K using LY294002. Int J Radiat Oncol Biol Phys. 2003 Jul 1;56(3):846-53. [ Links ]
73. Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001 May 15;61(10):3986-97. [ Links ]
74. Kim D, Dan HC, Park S, Yang L, Liu Q, Kaneko S, et al. AKT/PKB signaling mechanisms in cancer and chemoresistance. Front Biosci. 2005 Jan 1;10:975-87. [ Links ]
75. Stoica GE, Franke TF, Moroni M, Mueller S, Morgan E, Iann MC, et al. Effect of estradiol on estrogen receptor-alpha gene expression and activity can be modulated by the ErbB2/PI 3-K/Akt pathway. Oncogene. 2003 Sep 11;22(39):7998-8011. [ Links ]
76. Mendez P, Azcoitia I, Garcia-Segura LM. Estrogen receptor alpha forms estrogen-dependent multimolecular complexes with insulin-like growth factor receptor and phosphatidylinositol 3-kinase in the adult rat brain. Brain Res Mol Brain Res. 2003 Apr 10;112(1-2):170-6. [ Links ]
77. Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004 Aug;6(2):117-27. [ Links ]
78. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 Jan 7;100(1):57-70. [ Links ]
79. Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J, Szabo S, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004 Aug;3(8):772-5. [ Links ]
80. Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci USA. 2005 Jan 18;102(3):802-7. [ Links ]
81. Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004 Apr 23;304(5670):554. [ Links ]
82. Campbell IG, Russell SE, Choong DY, Montgomery KG, Ciavarella ML, Hooi CS, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004 Nov 1;64(21):7678-81. [ Links ]
83. Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C, et al. PIK3CA is implicated as an oncogene in ovarian cancer. Nat Genet. 1999 Jan;21(1):99-102. [ Links ]
84. Woenckhaus J, Steger K, Werner E, Fenic I, Gamerdinger U, Dreyer T, et al. Genomic gain of PIK3CA and increased expression of p110alpha are associated with progression of dysplasia into invasive squamous cell carcinoma. J Pathol. 2002 Nov;198(3):335-42. [ Links ]
85. Ma YY, Wei SJ, Lin YC, Lung JC, Chang TC, Whang-Peng J, et al. PIK3CA as an oncogene in cervical cancer. Oncogene. 2000 May 25;19(23):2739-44. [ Links ]
86. Mizoguchi M, Nutt CL, Mohapatra G, Louis DN. Genetic alterations of phosphoinositide 3-kinase subunit genes in human glioblastomas. Brain Pathol. 2004 Oct;14(4):372-7. [ Links ]
87. Broderick DK, Di C, Parrett TJ, Samuels YR, Cummins JM, McLendon RE, et al. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004 Aug 1;64(15):5048-50. [ Links ]
88. Jucker M, Sudel K, Horn S, Sickel M, Wegner W, Fiedler W, et al. Expression of a mutated form of the p85alpha regulatory subunit of phosphatidylinositol 3-kinase in a Hodgkins lymphoma-derived cell line (CO). Leukemia. 2002 May;16(5):894-901. [ Links ]
89. Cheng JQ, Ruggeri B, Klein WM, Sonoda G, Altomare DA, Watson DK, et al. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc Natl Acad Sci USA. 1996 Apr 16;93(8):3636-41. [ Links ]
90. Ruggeri BA, Huang L, Wood M, Cheng JQ, Testa JR. Amplification and overexpression of the AKT2 oncogene in a subset of human pancreatic ductal adenocarcinomas. Mol Carcinog. 1998 Feb;21(2):81-6. [ Links ]
91. Bellacosa A, de Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995 Aug 22;64(4):280-5. [ Links ]
92. Staal SP. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2:amplification of AKT1 in a primary human gastric adenocarcinoma. Proc Natl Acad Sci USA. 1987 Jul;84(14):5034-7. [ Links ]
93. Eng C. PTEN:one gene, many syndromes. Hum Mutat. 2003 Sep;22(3):183-98. [ Links ]
94. Li YL, Tian Z, Wu DY, Fu BY, Xin Y. Loss of heterozygosity on 10q23.3 and mutation of tumor suppressor gene PTEN in gastric cancer and precancerous lesions. World J Gastroenterol. 2005 Jan 14;11(2):285-8. [ Links ]
95. Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997 Mar 28;275(5308):1943-7. [ Links ]
96. Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, et al. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997 Nov 15;57(22):4997-5000. [ Links ]
97. Garcia JM, Silva JM, Dominguez G, Gonzalez R, Navarro A, Carretero L, et al. Allelic loss of the PTEN region (10q23) in breast carcinomas of poor pathophenotype. Breast Cancer Res Treat. 1999 Oct;57(3):237-43. [ Links ]
98. Khan S, Kumagai T, Vora J, Bose N, Sehgal I, Koeffler PH, et al. PTEN promoter is methylated in a proportion of invasive breast cancers. Int J Cancer. 2004 Nov 10;112(3):407-10. [ Links ]
99. Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, Wasserman L, et al. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res. 2004 May 1;64(9):3014-21. [ Links ]
100. Stephens P, Edkins S, Davies H, Greenman C, Cox C, Hunter C, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet. 2005 Jun;37(6):590-2. [ Links ]
101. Davies H, Hunter C, Smith R, Stephens P, Greenman C, Bignell G, et al. Somatic mutations of the protein kinase gene family in human lung cancer. Cancer Res. 2005 Sep 1;65(17):7591-5. [ Links ]
102. Parsons DW, Wang TL, Samuels Y, Bardelli A, Cummins JM, DeLong L, et al. Colorectal cancer:mutations in a signalling pathway. Nature. 2005 Aug 11;436(7052):792. [ Links ]
103. Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004 May 21;304(5674):1164-6. [ Links ]
104. Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, et al. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997 May 2;89(3):457-67. [ Links ]
105. Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988 May 20;53(4):549-54. [ Links ]
106. Yanez L, Groffen J, Valenzuela DM. c-K-ras mutations in human carcinomas occur preferentially in codon 12. Oncogene. 1987;1(3):315-8. [ Links ]
107. Nelson MA, Wymer J, Clements N, Jr. Detection of K-ras gene mutations in non-neoplastic lung tissue and lung cancers. Cancer Lett. 1996 May 15;103(1):115-21. [ Links ]
108. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002 Jun 27;417(6892):949-54. [ Links ]
109. Rolitsky CD, Theil KS, McGaughy VR, Copeland LJ, Niemann TH. HER-2/neu amplification and overexpression in endometrial carcinoma. Int J Gynecol Pathol. 1999 Apr;18(2):138-43. [ Links ]
110. Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu protooncogene in human breast and ovarian cancer. Science. 1989 May 12;244(4905):707-12. [ Links ]
111. Stephens P, Hunter C, Bignell G, Edkins S, Davies H, Teague J, et al. Lung cancer:intragenic ERBB2 kinase mutations in tumours. Nature. 2004 Sep 30;431(7008):525-6. [ Links ]
112. Klos KS, Zhou X, Lee S, Zhang L, Yang W, Nagata Y, et al. Combined trastuzumab and paclitaxel treatment better inhibits ErbB-2-mediated angiogenesis in breast carcinoma through a more effective inhibition of Akt than either treatment alone. Cancer. 2003 Oct 1;98(7):1377-85. [ Links ]
113. Nicholson KM, Streuli CH, Anderson NG. Autocrine signalling through erbB receptors promotes constitutive activation of protein kinase B/Akt in breast cancer cell lines. Breast Cancer Res Treat. 2003 Sep;81(2):117-28. [ Links ]
114.Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci USA. 1992 May 15;89(10):4309-13. [ Links ]
115. Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004 Aug 20;305(5687):1163-7. [ Links ]
116. Tang CK, Gong XQ, Moscatello DK, Wong AJ, Lippman ME. Epidermal growth factor receptor vIII enhances tumorigenicity in human breast cancer. Cancer Res. 2000 Jun 1;60(11):3081-7. [ Links ]
117. Barber TD, Vogelstein B, Kinzler KW, Velculescu VE. Somatic mutations of EGFR in colorectal cancers and glioblastomas. N Engl J Med. 2004 Dec 30;351(27):2883. [ Links ]
118. Okamoto I, Kenyon LC, Emlet DR, Mori T, Sasaki J, Hirosako S, et al. Expression of constitutively activated EGFRvIII in non-small cell lung cancer. Cancer Sci. 2003 Jan;94(1):50-6. [ Links ]
119. Rosenzweig KE, Youmell MB, Palayoor ST, Price BD. Radiosensitization of human tumor cells by the phosphatidylinositol3-kinase inhibitors wortmannin and LY294002 correlates with inhibition of DNAdependent protein kinase and prolonged G2-M delay. Clin Cancer Res. 1997 Jul;3(7):1149-56. [ Links ]
120. Ng SS, Tsao MS, Nicklee T, Hedley DW. Wortmannin inhibits pkb/akt phosphorylation and promotes gemcitabine antitumor activity in orthotopic human pancreatic cancer xenografts in immunodeficient mice. Clin Cancer Res. 2001 Oct;7(10):3269-75. [ Links ]

