










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Ciencias de la Salud
versión impresa ISSN 1692-7273versión On-line ISSN 2145-4507
Rev. Cienc. Salud v.10 n.1 Bogotá ene./abr. 2012
Pérdida de heterocigocidad e identificación de portadoras de distrofia muscular de Duchenne: un caso familiar con evento de recombinación*
Loss of heterozygosity and carrier identification in Duchenne muscular dystrophy: a familiar case with recombination event
Perda de heterozigosidade e identificação de portadoras de distrofia muscular de Duchenne: um caso familiar com evento de recombinação
Dora Janeth Fonseca Mendoza, MSc1, Heidi Mateus Arbeláez, MSc1, Claudia Tamar Silva Aldana, MSc1
* Este trabajo fue presentado en modalidad de Presentación Oral en el V Congreso Colombiano de Genética, octubre de 2010. Los resultados expuestos en este trabajo forman parte de los análisis hechos en el proyecto "Análisis de dosis génica y construcción de haplotipos para la caracterización de portadoras de mutaciones en el gen de la distrofina", financiado por Colciencias según el código 1222-04-16333.
1Unidad de Genética, Escuela de Medicina y Ciencias de la Salud, Universidad del Rosario. Bogotá-Colombia. Correspondencia: dora.fonseca@urosario.edu.co
Recibido: junio 21 de 2011 • Aprobado: febrero 16 de 2012
Para citar este artículo: Fonseca DJ, Mateus H, Silvia CT. Perdida de heterocigocidad e identificación de portadoras de distrofia muscular de Duchenne: un caso familiar con evento de recombinación. Rev. Cienc. Salud 2012; 10 (1):83-90.
Resumen
La distrofia muscular de Duchenne y Becker (DMD/DMB) es una entidad de herencia recesiva ligada al cromosoma X que se presenta con debilidad muscular y es causada por mutaciones en el gen de la distrofina. La pérdida de heterocigocidad permite identificar a las mujeres portadoras de deleción en el gen de la distrofina mediante haplotipos. Objetivo: identificar mujeres portadoras en una familia con un paciente afectado por DMD mediante análisis de pérdida de heterocigocidad. Materiales y métodos: se analizaron nueve miembros de una familia con un afectado de DMD. Se hizo extracción de ADN y amplificación de diez STR del gen de la distrofina; se construyeron haplotipos, y se determinó el estado de portadora de deleción en dos de las seis mujeres analizadas, quienes mostraron pérdida de heterocigocidad de tres STR. Se establecieron algunos eventos de recombinación. Resultados: dos de las seis mujeres analizadas, mostraron pérdida de heterocigocidad en tres de los diez STR genotipificados, indicando su estado de portadora de deleción en este fragmento del gen de la distrofina. Con la segregación familiar de los haplotipos se establecieron eventos de recombinación. Conclusiones: mediante pérdida de heterocigocidad es posible establecer el estado de portadora de deleción en el gen de la distrofina con un 100% de certeza. La construcción de haplotipos identifica el cromosoma X portador de la deleción en familiares del caso índice. Se evidenció un evento de recombinación en una de las hermanas del afectado, lo que hace indeterminado su estado de portadora.
Palabras clave: recombinación, pérdida de heterocigocidad, distrofina, mosaicismo.
Abstract
Duchenne/Becker Muscular Dystrophy (DMD/BMD) is an X-linked recessive disease characterized by muscular weakness. It is caused by mutations on the dystrophin gen. Loss of heterozygosity allows us to identify female carriers of deletions on the dystrophin gen. Objective: identify female carriers in a family with a patient affected by DMD. Material and methods: nine family members and the affected child were analyzed using DNA extraction and posterior amplification of ten STRs on the dystrophin gen. Haplotypes were constructed and the carrier status determined in two of the six women analyzed due to loss of heterozygosity in three STRs. Additionally, we observed a recombination event. Conclusions: loss of heterozygosity allows us to establish with a certainty of 100% the carrier status of females with deletions on the dystrophin gen. By the construction of haplotypes we were able to identify the X chromosome with the deletion in two of the six women analyzed. We also determined a recombination event in one of the sisters of the affected child. These are described with a high frequency (12%). A possible origin for the mutation is a gonadal mosaicism in the maternal grandfather or in the mother of the affected child in a very early stage in embryogensis. This can be concluded using the analysis of haplotypes.
Keywords: recombination, loss of heterozygosity, dystrophin, mosaicism.
Resumo
A distrofia muscular de Duchenne e Becker (DMD/DMB) é uma entidade de herança recessiva ligada ao cromossoma X que se apresenta com debilidade muscular e é causada por mutações no gene da distrofia. A perda de heterozigosidade permite identificar às mulheres portadoras de deleção no gene da distrofina mediante haplótipos. Objetivo: identificar mulheres portadoras em uma família com um paciente afetado de DMD mediante análises de perda de heterozigosidade. Materiais e métodos: se analisaram nove membros de uma família com um afetado de DMD. De fez extração de ADN e amplificação de dez STR do gene da distrofina; construíram-se haplótipos, e determinou-se o estado de portadora de deleção em duas das seis mulheres analisadas, as quais mostraram perda de heterozigosidade de três STR. Estabeleceram-se alguns eventos de recombinação. Resultados: duas das seis mulheres analisadas mostraram perda de heterozigosidade em três dos dez STR genotipados, indicando seu estado de portadora de deleção neste fragmento do gene da distrofina. Com a segregação familiar dos haplótipos se estabeleceram eventos de recombinação. Conclusões: mediante perda de heterozigosidade é possível estabelecer o estado de portadora de deleção no gene da distrofina com um 100% de certeza. A construção de haplótipos identifica o cromossoma X portador da deleção em familiares do caso índice. Evidenciou-se um evento de recombinação em uma das irmãs do afetado, o que faz indeterminado seu estado de portadora.
Palavras chave: recombinação, perda de heterozigosidade, distrofina, mosaicismo.
La distrofia muscular de Ducehenne (DMD) y su forma alélica menos severa, la distrofia muscular de Becker (DMB), son enfermedades de herencia recesiva ligada al cromosoma X (1). La DMD afecta a uno de cada 3.500 niños nacidos vivos, mientras la DMB afecta a uno de cada 7.500 (2); estas dos entidades genéticas se deben a mutaciones en el gen de la distrofina localizado en Xp21, el cual codifica para la proteína de la membrana del citoesqueleto en el músculo esquelético. El daño del gen resulta en degeneración muscular progresiva y muerte temprana. Las mutaciones que causan la enfermedad son duplicaciones, mutaciones puntuales y deleciones (de uno o varios exones), siendo estas las más frecuentes (3). Nuestro grupo de investigación ha encontrado que en Colombia la frecuencia de pacientes con deleciones es cerca de 32% (4-5), aunque otros estudios han reportado valores hasta de 98% (6-7). La mayoría de las deleciones afectan exones localizados dentro de dos regiones denominadas puntos calientes. Cerca de un tercio de los pacientes tiene mutaciones de nuevo; sin embargo, en algunas poblaciones se han reportado tasas hasta de 66% (8).
El gen de la distrofina tiene 2,4 megabases y es uno de los más grandes descritos hasta el momento; contiene 69 exones y se ha planteado que su gran tamaño tiene una estrecha relación con su alta tasa de mutaciones (6, 9). La identificación de las deleciones en afectados se realiza usualmente mediante reacción en cadena de la polimerasa, PCR, múltiplex (10); no obstante, con esta misma metodología la determinación del estado de portadora es más difícil debido a que al tener dos cromosomas X, la presencia del alelo normal enmascara la deleción del X anormal (11). En los últimos años se han implementado estrategias de análisis indirecto mediante construcción de haplotipos, usando repeticiones cortas en tándem (STR por su nombre en inglés Short Tandem Repeats) que permiten establecer el cromosoma X ligado con la enfermedad, estrategia que reconoce portadoras en aquellos casos en que la mutación del afectado no ha sido registrada (12-13).
Por otro lado, la construcción de haplotipos con STR puede convertirse en un análisis directo en casos donde la mutación del afectado es conocida y corresponde a una deleción dentro de la región analizada. Esta es una estrategia alternativa que permite asignar estado de portadora mediante el registro de pérdida de heterocigocidad en uno o varios de los STR involucrados en la deleción (14).
Se realizó la construcción de haplotipos con STR situados en el gen de la distrofina en nueve personas de dos generaciones pertenecientes a una familia con un afectado por DMD, en quien nuestro grupo detectó una deleción de los exones 48 al 52. El análisis del haplotipo indicó pérdida de heterocigocidad para los sistemas DXS1236, DXS1235 y DXS1036 en dos de las mujeres analizadas.
Materiales y métodos
Previo consentimiento informado se tomaron muestras de sangre periférica anticoagulada con EDTA en nueve miembros de la familia de un afectado por DMD (Figura 1), en quien se había determinado la deleción de parte del gen. Se hizo extracción de ADN y posterior amplificación mediante PCR de diez microsatélites o STR (CA)n situados a lo largo del gen de la distrofina (Tabla 1). Dos de los marcadores analizados, DXS1242 y DXS992, fueron extragénicos y los demás fueron intragénicos: STR-2, DXS205, DXS1219, DXS1237, DXS1236, DXS1235, DXS1036 y DXS1214. La reacción de PCR fue realizada bajo condiciones estandarizadas y descritas por nuestro grupo de investigación con anterioridad (13).
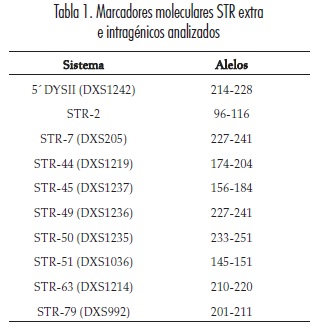
Los productos amplificados fueron sembrados sobre geles de secuenciación y corridos en electroforesis vertical durante 200 minutos a 1.500V, 60 mA y 50 °C en el secuenciador automático ALF Express (Pharmacia). Mediante las aplicaciones ALFWin y Allelinks® fueron asignados los alelos de cada STR según el tamaño en pares de bases de los productos obtenidos para cada individuo analizado, teniendo como control de asignación los pesos reportados en la literatura para cada alelo. En cada corrido se usaron marcadores externos (50-500 pb) e internos (250 o 300 pb) que permitían asignar de manera inequívoca el alelo de cada persona. La información alélica fue organizada y analizada para cada uno de los miembros de la familia y se realizó la construcción del haplotipo, partiendo de que el construido para el afectado es el ligado a la mutación. La asignación de pérdida de heterocigocidad se estableció después de tres corridos que no evidenciarán señal de amplificación para un mismo sistema. Los eventos de recombinación fueron identificados mediante comparación de los haplotipos de la madre y hermanas del afectado, de tal manera que cualquier cambio de la información alélica del haplotipo se consideró una recombinación.
Resultados
La construcción de haplotipos mediante los STR analizados en el paciente afectado con DMD permitió señalar el cromosoma X que en esta familia está ligado a la enfermedad (XA) (Figuras 1 y 2) e indica una deleción que involucra los STR DXS1236, DXS1235 y DXS1036, situados en los intrones 49, 50 y 51 del gen de la distrofina. En dos de las mujeres analizadas, que corresponden a la madre (I3) y una de las hermanas (II1) del afectado (Figuras 1), se identificó este mismo cromosoma XA que indica su estado de portadora de deleción en este segmento del gen de la distrofina. En el resto de las mujeres familiares del afectado no se heredó el cromosoma ligado a la enfermedad, por lo que se consideran no portadoras y sin riesgo de tener hijos afectados de DMD.

En la hermana portadora del afectado (II1) se mostró pérdida de heterocigocidad para los sistemas DXS1236, DXS1235 y DXS1036 mediante los análisis de electroforetogramas que indican amplificación solo para el alelo heredado del padre ya que de la madre ha recibido este segmento delecionado y por lo tanto sin ningún alelo del STR detectable por PCR (Figura 2).
En una de las hermanas no portadoras (II4) se identificó un evento de recombinación entre los dos cromosomas XA y XB maternos en el sistema STR-2, demostrado por la presencia de un alelo 118 y no el 120 esperado (Figuras 1).
La información del haplotipo materno heredado (XB) a la hija con la recombinación (II4) permite inferir que la extensión de la recombinación no fue más allá del STR DXS1219, situado en el intrón 44 del gen de la distrofina y por lo tanto no involucró el segmento que porta la deleción presente en el afectado (II5) y su hermana portadora que cubre desde el intrón 49 hasta el 51 (Figuras 1).
El estudio conjunto de las tías maternas (I1 y I2) y la madre del afectado (I3) permitió concluir que el cromosoma XA ligado con la enfermedad fue heredado del abuelo materno.
Discusión
El análisis del estado de portadora en DMD, enfermedad recesiva ligada al cromosoma X, ha sido investigado con amplitud ya que constituye la única manera de realizar prevención primaria de esta entidad de gran morbimortalidad y aún sin terapia efectiva descrita (12, 15). Para lograr este objetivo se han señalado diversas estrategias como el southern blot, la densitometría y el análisis de dosis génica mediante PCR en tiempo real. La construcción de haplotipos mediante marcadores altamente polimórficos como STR ha sido muy utilizado como método indirecto para reconocer portadoras, por medio del rastreo del cromosoma X ligado con la enfermedad; sin embargo, en casos como el planteado en este artículo, la presencia de deleciones que involucren uno o varios de los STR analizados y que lleven a pérdida de la heterocigocidad logra establecer el estado de portadora con una sensibilidad de 100%. El haplotipo de riesgo, que es compartido con el afectado, permite rastrear a las mujeres por línea materna que tendrán riesgo de 50% de tener hijos varones afectados de DMD, lo que permitirá explicar opciones reproductivas alternativas, como donación de óvulos o diagnóstico pre implantación, entre otros, mediante asesoramiento genético.
La pérdida de heterocigocidad se observa como una falsa homocigocidad para uno o varios STR situados en la región delecionada. Así, mujeres que deberían ser heterocigotas obligadas (alelo materno y paterno diferente) solo exhiben la presencia de uno de sus dos alelos. Dado el tipo de herencia recesiva ligada a X de la enfermedad, el alelo normal es heredado por el padre, mientras la madre hereda su alelo con la mutación de tipo deleción, que señala el estado de portadora percibido en la mujer II-3 (Figuras 1).(Figura 2).
Se ha descrito que el gen de la distrofina sufre de una alta tasa de eventos de recombinación (crossing over) durante la meiosis debido a la alta homología entre diferentes secuencias dentro de este (16). Estos eventos son detectables con facilidad por la comparación de los haplotipos entre madres e hijas, ya que se aprecian cambios de la información alélica esperada. En una de las hermanas del afectado (II4) se observó este evento, en el cual un STR situado en el intrón 2 del gen fue involucrado en el intercambio entre los cromosomas XA y XB maternos (Figura 1). La extensión de la recombinación no involucró la región delecionada del gen (intrones 49 a 51), lo que permite descartar a esta paciente como portadora. La determinación de la extensión de la recombinación se puede lograr solo si la informatividad de los sistemas STR utilizados lo permiten; en casos donde los alelos sobre los dos cromosomas X son iguales (mujeres homocigotas) no se podría identificar si hubo intercambio o no. Así, respecto a la mujer descrita en este trabajo podemos inferir que la recombinación involucró como mínimo el STR 2 y como máximo el STR 44.
En Colombia se ha descrito que la heterocigocidad para los STR analizados está entre 35,1 y 81,1%, lo cual los hace altamente polimórficos y por lo tanto útiles para este tipo de análisis (13). En análisis indirectos por análisis de haplotipos, una recombinación entre el marcador utilizado y el exón de interés puede causar errores diagnósticos, ya que una mujer portadora con el haplotipo de riesgo podría no tener la mutación en el exón de interés o, por el contrario, se puede poseer el haplotipo no ligado a la mutación y portar la enfermedad, situación que obliga a establecer como indeterminado el estado o no de portadora en las mujeres con evidencia de recombinación (17-18).
En la mujer I3 analizada se reveló una pérdida de heterocigocidad sobre el cromosoma X heredado del padre (Figura 1). Se esperaría que las mujeres I1 y I2 mostraran esta misma condición, pero en ellas no se presentó ninguna deleción. Esta observación permite plantear una hipótesis de un evento mutacional de novo en la madre del afectado (mosaicismo somático) o en las células germinales del abuelo materno del paciente (mosaicismo germinal); sin embargo, los resultados del presente estudio no permiten demostrar cuál de estas dos posibilidades biológicas precisó el estado de portadora de la paciente I3.
Cerca de un tercio de las mutaciones que llevan a DMD/DMB corresponden a mutaciones de novo y dentro de ellas las deleciones de novo son frecuentes, en especial en casos de DMD esporádicos o aislado, con valores que llegan a un 62,2% (19).
Conclusión
En este artículo se describió una familia con un afectado de DMD que presenta deleción de los exones 48 al 52. A pesar de ser un caso esporádico, la precisión del estado de portadora de sus hermanas y madre fue posible por la identificación de pérdida de heterocigocidad en los STR que se involucraron en la zona delecionada. Se analiza también la presencia y consecuencias del evento de recombinación percibido en la hermana II-3, así como la posibilidad que el origen de la mutación para esta familia es un mosaicismo germinal del abuelo materno o una mutación de novo universal en la madre. El reconocimiento de portadoras de DMD mediante pérdida de heterocigocidad es una estrategia alternativa que permite ofrecer asesoramiento genético adecuado y logra establecer con 100% de sensibilidad el riesgo de tener hijos varones afectados.
El análisis de segregación familiar mediante marcadores STR se convierte en una herramienta molecular bastante eficiente en la detección molecular de portadoras y el diagnóstico prenatal de DMD/DMB (12, 15, 20).
Agradecimientos
Los autores expresan su agradecimiento a la familia descrita en el presente artículo, a la señora Lida Triviño por su ayuda en algunos procesos de laboratorio, así mismo a la Universidad del Rosario y Colciencias por la financiación de este trabajo.
Descargos de responsabilidad
Los autores manifiestan que no existe ningún conflicto de interés que afecte los resultados y los análisis consignados en el texto de este artículo.
Bibliografía
1. Worton RG, Burghes AH. Molecular gentics of Duchenne and Becker muscular dystrophy. Int Rev Neurobiol. 1988; 29:1-76. [ Links ]
2. Emery AE, Skinner R, Holloway S. A study of possible heterogeneity in Duchenne muscular dystrophy. Clin Gent. 1979; 1 (5):444-9. [ Links ]
3. Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987; 51 (6):919-28. [ Links ]
4. Silva CT, Fonseca D, Restrepo CM, Contreras N, Mateus H. Deleciones en el gen de la distrofina en 62 familias colombianas: correlación genotipo-fenotipo para la distrofina muscular de Duchenne y Becker. Colom. Med. 2004; 35 (4):191-8. [ Links ]
5. Silva CT, Mateus H, Fonseca DJ, Simbaqueba J. Identificación de deleciones en exones situados fuera del "Hot Spot" distal del gen de la distrofina en pacientes afectados con distrofia muscular de Duchenne. Revista Cuadernos de Medicina en Investigación y Salud 2007; 1 (3):206-12. [ Links ]
6. Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987; 50 (3):509-17. [ Links ]
7. Beggs AH, Koenig M, Boyce FM, Kunkel LM. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum. Genet. 1990; 86 (1):45-8. [ Links ]
8. Sinha S, Mishra S, Singh V, Mittal RD, Mittal B. High frequency of new mutations in North Indian Duchenne/Becker muscular dystrophy patients. Clin Gent. 1996; 50 (5):327-31. [ Links ]
9. Van Ommen GJ, Bertelson C, Ginjaar HB, den Dunnen JT, Bakker E, Chelly J et al. Long-range genomic map of the Duchenne muscular dystrophy (DMD) gene: isolation and use of J66 (DXS268), a distal intragenic marker. Genomics 1987; 1 (4):329-36. [ Links ]
10. Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. 1988; 16 (23):11141-56. [ Links ]
11. Bunyan DJ, Robinson DO, Collins AL, Cockwell AE, Bullman HM, Whittaker PA. Germline and somatic mosaicism in a female carrier of Duchenne muscular dystrophy. Hum. Genet. 1994; 93 (5):541-4. [ Links ]
12. Giliberto F, Ferreiro V, Massot F, Ferrer M, Francipane L, Szijan I. Prenatal Diagnosis of Duchenne/ Becker muscular dystrophy by short tandem repeat segregation analysis in Argentine families. Muscle Nerve 2011; 43 (4):510-17. [ Links ]
13. Fonseca D, Silva C, Mateus H. Detección de portadoras de distrofia muscular de Duchenne en familias colombianas mediante análisis de microsatélites. Colom. Med. 2008a; 39 (2): 7-13. [ Links ]
14. Fonseca D, Silva C, Mateus H, Restrepo C. Identificación de deleciones en portadoras de distrofia muscular de Duchenne. Acta Colom. Med. 2008b; 33 (2):63-7. [ Links ]
15. Helderman-van den Enden AT, van den Bergen JC, Breuning MH, Verschuuren JJ, Tibben A, Bakker E et al. Duchenne/Becker muscular dystrophy in the familiy: have potential carriers been tested at a molecular level? Clin. Genet. 2011; 79 (3):236-42. [ Links ]
16. Chavarría G, Reis A, Azofeifa J. Determinación indirecta, mediante marcadores de ADN, del estado de portadoras de distrofia muscular de Duchenne (DMD) en una familia costarricense. Acta pediátr. costarric. 2002; 16 (1):2-38. [ Links ]
17. Carsana A, Frisso G, Tremolaterra MR, Ricci E, De Rasmo D, Salvatore F. A larger spectrum of intragenic short tandem repeats improves linkage analysis and localization of intragenic recombination detection in the dystrophin gene: an analysis of 93 families from Southern Italy. J. Mol. Diagn. 2007; 9 (1):64-9. [ Links ]
18. Alcántara MA, Villarreal MT, Del Castillo V, Gutiérrez G, Saldaña Y, Maulen I et al. High frequency of de novo deletions in Mexican Duchenne and Becker muscular dystrophy patients. Implications for genetic counseling. Clin. Genet. 1999; 55 (5):376-80. [ Links ]
19. Basak J, Dasgupta UB, Mukherjee SC, Das SK, Senapati AK, Banerjee TK. Deletional mutations of dystrophin gene and carrier detection in Eastern India. Indian J. Pediatr. 2009; 76 (10):1007-12. [ Links ]

