










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Ciencias de la Salud
versión impresa ISSN 1692-7273
Rev. Cienc. Salud vol.10 no.3 Bogotá sep./dic. 2012
Mecanismos moleculares involucrados en la mutagenicidad
inducida por aflatoxina B1
Molecular mechanism involved in the mutagenicity induced by Aflatoxin B1
Mecanismos moleculares envolvidos na mutagenicidade induzida pela aflatoxina B1
Diego Fernando Uribe-Yunda, Biol1, María-Cristina Navas MSc, PhD1
1 Grupo de Gastrohepatología, Facultad de Medicina, SIU, Universidad de Antioquia. Medellín. Correspondencia: mcnvasn@gmail.com
Recibido: septiembre 20 de 2011 • Aprobado: octubre 4 de 2012
Para citar este artículo: Uribe-Yunda DF, Navas MC. Mecanismos moleculares involucrados en la mutagenicidad inducida por aflatoxina B1. Rev Cienc Salud 2012; 10 (3): 403-419.
Resumen
La aflatoxina B1 (AFB1) es una micotoxina identificada como el más potente hepatocarcinógeno. El metabolito que resulta del proceso de detoxificación de la AFB1 en el hígado tiene la capacidad de reaccionar con el ADN genómico, generando el aducto AFB1-ADN; durante la replicación del ADN este aducto induce la transversión G:C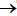 T:A. Polimorfismos en los genes que codifican las enzimas encargadas de la activación y detoxificación de la AFB1 y enzimas de reparación del ADN han sido asociados con el riesgo de desarrollar carcinoma hepatocelular (CHC). Adicionalmente, en poblaciones con alta exposición a aflatoxina y alta prevalencia de infección por el virus de la hepatitis B (VHB) se ha demostrado un sinergismo entre estos dos factores de riesgo para el desarrollo de CHC.
T:A. Polimorfismos en los genes que codifican las enzimas encargadas de la activación y detoxificación de la AFB1 y enzimas de reparación del ADN han sido asociados con el riesgo de desarrollar carcinoma hepatocelular (CHC). Adicionalmente, en poblaciones con alta exposición a aflatoxina y alta prevalencia de infección por el virus de la hepatitis B (VHB) se ha demostrado un sinergismo entre estos dos factores de riesgo para el desarrollo de CHC.
Palabras clave: aflatoxina B1, VHB, carcinoma hepatocelular, metabolito AFB1, Aductos AFB1-ADN, reparación del ADN.
Abstract
The aflatoxin B1 (AFB1) is a mycotoxin that has been identified as the most potent hepatocarcinogen. The metabolite resulting from detoxification process of AFB1 in liver, has the ability to react with the genomic DNA, generating AFB1-DNA adducts; during DNA replication process, this adduct induced the G:CT:A transversion. Polymorphism in genes encoding for enzymes involved in the activation and detoxification of AFB1 and DNA repair enzymes has been associated with the risk of hepatocellular carcinoma (HCC) development. Additionally, in populations of high exposure to aflatoxin and high prevalence of hepatitis B virus (HBV) infection, has been demonstrated a synergism between these two risk factors for the development of HCC.
Keywords: Aflatoxin B1, Hepatitis B virus, Hepatocellular carcinoma, AFB1 metabolite, AFB1- DNA adducts, DNA repair.
Resumo
Aflatoxina B1 (AFB1) é uma micotoxina identificado como o hepatocarcinogen mais potente. O metabolito resultante do processo de desintoxicação de AFB1 no fígado, tem a capacidade de reagir com o ADN genómico, gerando AFB1 DNA-aducto; transversão durante a replicação do ADN deste aducto induz G:CT:A. Polimorfismos em genes que codificam as enzimas envolvidas na activação e na desintoxicação de AFB1 e enzimas de reparação do ADN têm sido associados com o risco de desenvolvimento de carcinoma hepatocelular (HCC). Além disso, em populações com elevada exposição a aflatoxina e elevada prevalência da infecção com vírus da hepatite B (VHB) tem sido mostrado um sinergismo entre estes dois factores de risco para o desenvolvimento de carcinoma hepatocelular.
Palavras chave: aflatoxina B1, HBV, HCC, AFB1 metabolito, AFB1-DNA adutos, reparação do ADN.
Las aflatoxinas son metabolitos secundarios producidos por hongos del género Aspergillus que forman parte de la microflora del aire, principalmente Aspergillus flavus y Aspergillus parasiticus. Esta toxina puede estar presente en cereales como maíz, arroz, trigo y cebada y en frutos secos como maní y nueces, cuando estos son almacenados en condiciones inapropiadas de humedad y temperatura (1, 2). Este tipo de contaminación se presenta generalmente en regiones tropicales y semitropicales, en las cuales las condiciones de temperatura y humedad facilitan el crecimiento del hongo y la producción de las micotoxinas durante la mayor parte del año (3, 4).
La exposición a aflatoxina en humanos puede determinarse midiendo los grados de contaminación en alimentos y mediante la detección de biomarcadores en muestras de suero, orina y/o tejido hepático. La exposición varía de acuerdo con la región geográfica, siendo algunos países de África, como Kenia, Mozambique, Suráfrica y Gambia y países asiáticos como China y Tailandia, donde se registran los grados más altos. En Estados Unidos y gran parte de Europa la exposición es baja (5). Por otro lado, se ha observado una correlación directa entre la exposición a aflatoxina y el riesgo de desarrollar carcinoma hepatocelular (CHC); esto explicaría en parte las tasas altas de incidencia de CHC que se registran en países como Gambia y China.
Diferentes aflatoxinas han sido descritas y caracterizadas; sin embargo, las cuatro principales son B1, B2, G1 y G2 (6). La aflatoxina B1 (AFB1) ha sido clasificada como carcinógeno tipo 1 por la Agencia Internacional de Investigación en Cáncer (IARC) y como el más potente hepatocarcinógeno teniendo en cuenta las evidencias epidemiológicas y experimentales. Una de las evidencias corresponde a la transversión G:CT:A en la tercera posición del codón 249 de TP53 y en menor proporción las transversiones G:CT:A y G:CC:G en codones adyacentes, en células HepG2 a AFB1 activada con enzimas microsomales hepáticas de rata (7, 8). Adicionalmente la exposición dietaría a AFB1 se ha relacionado con la mutación en el codón 249 del gen TP53 en casos de CHC (9-12).
TP53 es el gen con más alta frecuencia de mutación en cáncer humano; la gran mayoría de las mutaciones se localizan en la secuencia que codifica el dominio de la proteína p53 de unión al ADN (13). En CHC se han descrito mutaciones en TP53 hasta en 60% de los casos analizados (14-16). Específicamente se ha encontrado una mutación puntual en la tercera posición del codón 249 en el exón 7 del gen TP53, que corresponde a una transversión G:CT:A. Esta mutación origina el cambio del aminoácido arginina por serina (mutación 249ser), alterando el dominio de unión de la proteína p53 al ADN (8-12).
En esta revisión se discutirán algunos de los mecanismos moleculares más relevantes en la mutagenicidad inducida por la AFB1. En la primera parte, la discusión se centra en el metabolismo de la micotoxina, la forma como reacciona con el ADN después de su activación y la reparación de los aductos AFB1-ADN, reseñando algunos de los modelos para el estudio de estos mecanismos. Luego se describirán otros factores importantes como el sinergismo entre la AFB1 y la infección por el virus de la hepatitis B (VHB) en la inducción de la mutación 249ser y el riesgo de desarrollar CHC.
Metabolismo de AFB1
El hígado es el principal órgano en el cual se realiza la biotransformación de los xenobióticos por acción de las enzimas de fase I y fase II. En términos generales, las enzimas de fase I, principalmente la superfamilia del Citocromo P450 (CYP450), convierten los metabolitos en compuestos polares, lo que facilita su excreción directa o su reacción con las enzimas de fase II. Las enzimas de fase II (por ejemplo, GlutatiónS-transferasa y Epóxido Hidrolasa) catalizan reacciones de conjugación para su excreción biliar y renal. Sin embargo, algunos metabolitos de la fase I pueden reaccionar con diferentes biomoléculas, lo que genera inestabilidad en las mismas.
Activación
La AFB1 es transformada a su forma activa AFB1 exo-8,9- epóxido, por enzimas microsomales hepáticas como el CYP450. El compuesto activado tiene la capacidad de reaccionar con el nitrógeno 7 de las guaninas del ADN produciendo aductos 8,9-dihidro-8-(N7-guanidil)- 9-hidroxiaflatoxina B1 (AFB1-N7-Gua); este puede ser convertido en un sitio apurínico o en un anillo imidazol de la guanina, formando el aducto estable AFB1 Formamidopirimidina (AFB1-FAPY), que corresponde al aducto más mutagénico (1, 11) (figura 1). En el sistema de Escherichia coli se demostró que el aducto AFB1-FAPY induce hasta seis veces más la transversión G:CT:A, que el aducto AFB1-N7-Gua. Además, se demostró que el aducto AFB1-FAPY es un potente bloqueador de la replicación del ADN, lo cual explica en parte la mutagenicidad inducida por la AFB1 (11).
Aunque diferentes isoformas de CYP450 participan en la transformación de la AFB1 en exo-epóxido, las más importantes son CYP1A2, CYP3A5 y CYP3A4; esta última es la isoforma de CYP450 más abundante en el hígado humano. La actividad de estas producen otros derivados de la AFB1, como AFM1, AFQ1 y AFB1 endo-8,9-epóxido. Sin embargo, estos productos tienen menor potencial mutagénico puesto que AFM1 y AFQ1 son pobres sustratos para la epoxidación y, en el caso de AFB1 endo 8,9-epóxido, la reacción con guaninas es poco eficiente (17-19).
Polimorfismos
La asociación de algunos polimorfismos en los genes que codifican estas enzimas con el riesgo de desarrollar CHC ha sido explorada en poblaciones de China y Gambia. Los polimorfismos en la región promotora y en la región codificante de un gen pueden tener efecto en la expresión y/o actividad catalítica de las proteínas codificadas.
En una población China con exposición dietaría a AFB1 y alta prevalencia de infección por el Virus de la hepatitis, B Chen y otros autores examinaron la presencia de polimorfismos en el gen CYP1A2; se evaluó el polimorfismo CYP1A2*1C, que corresponde a GA en las posiciones -3860 y -3113 del promotor y el polimorfismo CYP1A2*1B, TC en la posición 5347 de la región codificante del gen. Este estudio demostró que individuos homocigóticos portadores del haplotipo -3860G/-3113G/5347C presentaban un riesgo mayor de desarrollar CHC comparado con el grupo control (odds ratio [OR] = 1,65; intervalo de confianza de 95% [CI]: 1,11-2,46; p = 0,014); además este riesgo fue incluso mayor en individuos fumadores (OR = 2,14; 95% CI: 1,213,80; p = 0,009) (20).
La asociación entre polimorfismo de genes que codifican para enzimas microsomales hepáticas y el riesgo de desarrollar CHC también ha sido evaluada en poblaciones con alta incidencia de cáncer primario de hígado y exposición dietaría a AFB, como es el caso de Gambia.
Tres polimorfismos del gen CYP3A5 fueron examinados en población de este país del occidente africano. CYP3A5*3 (6986 AG) que genera un sitio de "splicing" e introduce un codón de parada, CYP3A5*6 (14690 GA) que genera un sitio de "splicing" en el exón 7 y provoca la pérdida de este exón y CYP3A5*7 en el que la inserción de una T genera un codón de parada en el codón 348 (27131-27132insT) (21, 22). Los investigadores describieron cuatro haplotipos en las muestras analizadas: i) 6986A/14690G/27131 sin inserción de T, con alta expresión de la proteína, ii) 6986G/14690G/27131 sin inserción de T, con expresión reducida de la proteína, iii) 6986A/14690A/27131 sin inserción de T, con expresión disminuida de la proteína y iv) 6986A/14690G/27131-27132insT, con expresión disminuida de la proteína. Individuos homocigóticos para el haplotipo i presentaron un incremento de 23,2% en los niveles de aductos aflatoxina-albúmina (AF-alb), comparado con lo observado en muestras de individuos con los haplotipos ii, iii, iv, que se caracterizaban por un nivel de expresión disminuido de la enzima microsomal. El porcentaje de aductos se incrementó hasta un 70% en individuos con actividad disminuida de CYP3A4 (23).
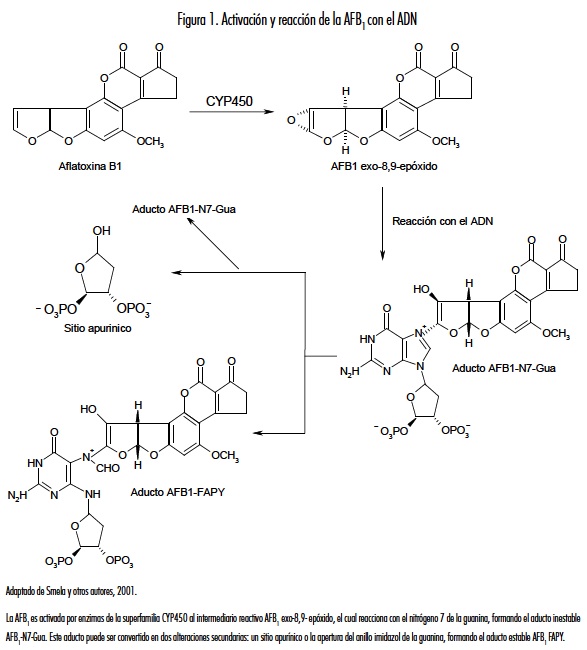
Detoxificación
La conjugación del metabolito intermediario AFB1 exo-8,9-epóxido con glutatión (GSH), mediada por Glutatión-S-transferasa (GST), es el principal mecanismo por el cual sucede la detoxificación de la AFB1 (24). También se ha postulado que la enzima epóxido hidrolasa (HYL1) participa en la hidrólisis de AFB1 exo-8,9-epóxido a aflatoxina-dihidrodíol; sin embargo, los resultados de los estudios son contradictorios (figura 2) (24, 25).
Diferentes isoformas de la proteína GST han sido caracterizadas, entre ellas glutatión-S-tranferasa p1 (GSTM1) y glutation-S-tranferasa 91 (GSTT1). Sun y otros autores evaluaron el polimorfismo en GSTM1 y GSTT1 y el riesgo de CHC en una población de Taiwan, país con alta prevalencia de infección por VHB. Los investigadores observaron que el riesgo de CHC era mayor en individuos con exposición a AFB1, marcador de infección por VHB (antígeno de superficie HBsAg) y GSTT1; una tendencia similar fue observada con GSTM1 aunque las diferencias no fueron estadísticamente significativas (26).
Estos resultados fueron parcialmente confirmados en un estudio similar realizado en población de Gambia, en el cual se observó una asociación entre GSTM1 y el riesgo de desarrollar CHC, pero no con GSTT1 (27).
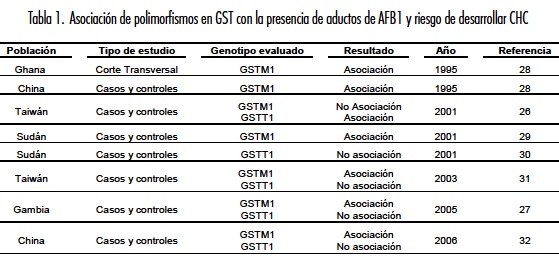
Estudios similares a los mencionados realizados en diferentes regiones del mundo confirman la importancia de GST en el metabolismo de la AFB1 y sugieren una relación de los polimorfismos GSTM1 y GSTT1 con el riesgo de desarrollar CHC (tabla 1) (28-32).
Como se dijo, la epóxido hidrolasa (HYL) participa en la hidrólisis de AFB1 exo-8,9-epóxido.
El polimorfismo HYL1*2 corresponde a una transición T:AC:G en el nucleótido 113 de este gen, da origen al cambio del aminoácido tirosina por histidina y está asociado con una disminución de 40% en la actividad in vitro de la enzima (33). Un estudio realizado en muestras de pacientes con CHC provenientes de dos regiones de alta exposición a AFB1 en Ghana y China permitió demostrar la asociación de la variante HYL1*2 con incremento en los niveles de aductos de AFB1 y el riesgo de CHC. Los pacientes que portaban este polimorfismo tenían niveles muy superiores de aductos AFalb, comparado con los controles (28). En otros estudios realizados en zonas de alta exposición a AFB1 en diferentes países, la variante HYL1*2 no fue un factor asociado con el desarrollo de CHC; sin embargo, al realizar análisis que incluían los polimorfismos en GST y la variante HYL1*2, fue observado un incremento significativo en el riesgo de CHC (27, 28, 30, 32, 34).
Reacción AFB1-ADN
Luego de la ingestión de alimentos contaminados con AFB1 se produce el metabolito AFB1 exo-8,9-epóxido gracias a la actividad de las enzimas microsomales hepáticas.
Este metabolito es considerado como el más potente hepatocarcinógeno por su capacidad de reaccionar con el ADN celular. Inicialmente AFB1 exo-8,9-epóxido se intercala en la doble cadena del ADN, específicamente en el extremo 5' de la guanina blanco; después, el par de electrones libres del nitrógeno 7 de la guanina atacan el carbono 8 del epóxido para formar el aducto AFB1-N7-Gua (1, 35). Algunos estudios sugieren que la reactividad de la AFB1 por la guanina depende de las bases contiguas de la guanina blanco tanto en el extremo 5' como en el 3'; la reactividad sería mayor en una secuencia de tres guaninas (GGG) (36).
Estudios in vitro con dúplex de oligonucleótidos correspondientes a secuencias del gen TP53 permitieron cuantificar los aductos formados luego de la exposición a AFB1 epóxido; esta estrategia permitió determinar la reactividad de los nucleótidos guaninas presentes en las secuencias en diferentes ubicaciones. En los ensayos con el oligonucleótido 5'-CCGGA-GGCC-3' (nucleótidos 741-749) del ADNc del gen TP53, se demostró que la guanina 747, que corresponde a la tercera posición del codón 249 de TP53, presenta una reactividad significativa (35). Estos resultados fueron confirmados en estudios posteriores, aportando evidencia de que el codón 249 de TP53 es un "hot spot" para la formación de aductos AFB1-ADN (37, 38).
Por otro lado, se ha sugerido que la formación de aductos AFB1-ADN está favorecida por la presencia de citosinas metiladas en secuencias ricas en guaninas y citosinas, conocidas como islas CpG. Es de anotar que más de 30% de las mutaciones en TP53 se ubican en islas CpG en los codones 157, 175, 245, 248 y 282; sin embargo, la frecuencia de mutaciones en estas secuencias varía según el tipo de tumor y el tejido. In vitro se ha demostrado que la metilación de citosinas en islas CpG en TP53 aumenta de 2 a 5 veces la afinidad de diferentes tipos de carcinógenos por estas secuencias, incluyendo el carcinógeno AFB1 exo-8,9-epóxido; estos hallazgos sugieren que las secuencias CpG son "hot spot" para las mutaciones por la unión preferencial de carcinógenos, con respecto a otras secuencias del gen (39, 40).
Reparación aductos AFB1-ADN
Los mecanismos de reparación son los responsables de mantener la integridad del ADN (41). Diferentes modelos han sido utilizados para explorar los mecanismos por los cuales se fijan los aductos y los sistemas de reparación que intervienen en la reparación de los mismos; estos modelos han permitido dilucidar que el principal mecanismo de reparación corresponde a la escisión de nucleótidos (NER). NER es el sistema de reparación más versátil para detectar los daños en el ADN ya que reconoce las lesiones que distorsionan su estructura e interfieren con el apareamiento de bases, la transcripción y la replicación; además, está encargado de reparar la mayoría de los daños ocasionados por factores ambientales (41, 42).
Se ha sugerido que la reparación de los aductos AFB1-FAPY es menos eficiente que la de AFB1-N7-Gua en mamíferos, puesto que estos últimos distorsionan más la estructura del ADN y por tanto son fácilmente reconocidos por las maquinarías de reparación.
Las evidencias experimentales del papel de NER en la reparación de secuencias de ADN que presenta aductos como AFB1-FAPY han sido aportadas por estudios en bacterias como Escherichia coli, en levaduras como Saccharomyces cerevisiae, en líneas celulares y en modelos murinos. En la tabla 2 se presentan las proteínas que participan en el sistema de reparación NER de E. coli y S. cerevisiae y sus equivalentes en el genoma humano.
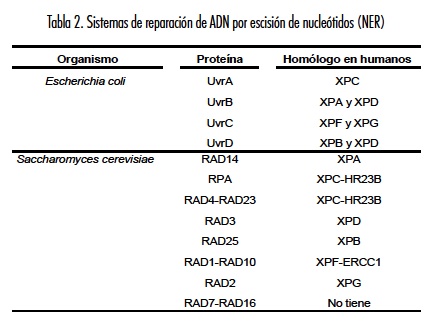
Utilizando cepas de E. coli, con diferente background genético con respecto a los sistemas de reparación de ADN, Alekseyev y otros autores demostraron que el aducto AFB1-FAPY era reparado preferencialmente por NER. En este sistema se demostró la remoción y reparación de los aductos presentes en un plásmido que contenía un gen reportero en bacterias wild type (wt), pero no en bacterias deficientes para NER (UvrA). En el caso de bacterias deficientes para el mecanismo por escisión de bases (mutM) se observó que la actividad del gen reportero fue similar a la descrita en bacterias wt; esto sugiere que la escisión de bases no cumple un papel importante en la reparación de los aductos AFB1-FAPY (43).
Estos resultados fueron confirmados por Guo y otros autores utilizando el modelo de S. cerevisiae. Levaduras deficientes en una de las proteínas del sistema NER, específicamente en la proteína Rad14, presentaron una viabilidad disminuida y alta frecuencia de mutaciones en respuesta al tratamiento con AFB1 (44).
NER también corresponde al sistema de elección para reparar el ADN por presencia de aductos como AFB1-FAPY en células de mamíferos. Esta aseveración fue demostrada utilizando fibroblastos humanos deficientes para xeroderma pigmentosum A (XPA); XPA es la proteína equivalente a UvrB y a RAD14 en los sistemas de reparación de E. coli y S. cerevisiae, respectivamente (tabla 2). La línea celular deficiente en XPA y un cultivo de fibroblastos normales fueron expuestos a AFB1, previo tratamiento de la micotoxina con enzimas microsomales hepáticas de rata (45).
La acumulación del aducto AFB1-N7-Gua fue evaluada en los dos tipos de cultivo celular; este aducto inestable fue removido a mayor velocidad en el cultivo de fibroblastos normales tanto por remoción espontánea como por los sistemas de reparación. En el caso de la línea celular deficiente para XPA la remoción fue menos eficiente. Con respecto a la acumulación del aducto AFB1-FAPY fue a mayor velocidad y en mayor proporción en la línea celular deficiente para XPA.
Resultados similares fueron obtenidos utilizando un plásmido de ADN modificado in vitro con AFB1 y transfectado en fibroblastos humanos deficientes para XPA (46). Estos estudios confirman la importancia del sistema NER en este tipo de reparación.
Por último, la frecuencia de tumores hepáticos en ratones deficientes para XPA por exposición a AFB1 aporta una evidencia muy importante del papel del sistema NER en condiciones de exposición a este carcinógeno (47).
Otros factores Infección por el virus de la hepatitis B
Estudios en poblaciones con exposición dietaría a AFB1 y alta prevalencia de infección por VHB aportaron las primeras evidencias del sinergismo entre estos dos factores de riesgo (48, 49).
Un individuo con exposición crónica a la AFB1 en su dieta, como es el caso de habitantes de algunos países africanos durante la estación de menor pluviosidad, tiene riesgo de desarrollar la mutación 249ser en TP53. Sin embargo, en un número importante de casos los sistemas de reparación del ADN, en particular NER, permitirán la remoción de los aductos y por tanto se evitará la fijación de la mutación 249ser.
Este no es el caso de individuos que además de estar expuestos a la AFB1 tienen infección crónica por VHB. En efecto se ha demostrado que este virus puede bloquear diferentes mecanismos de reparación del ADN.
Se ha demostrado que la proteína X del VHB (HBx) favorece la susceptibilidad a mutaciones gracias a su capacidad de unión a la proteína p53; de esta manera HBx inhibiría la actividad transactivadora de p53 sobre los promotores de genes involucrados en la reparación del ADN, apoptosis y en la regulación del ciclo celular.
Asimismo, se ha evidenciado que la reparación por NER, mecanismo por el cual se reparan preferencialmente los aductos AFB1-ADN, también se ve afectada (11, 43, 50, 51). Algunos estudios han comprobado la interacción de HBx con moléculas implicadas en el sistema NER acoplado a la transcripción, como XPB, XPD (componentes del factor TFIIH) y TFIIF y su unión directa al ADN dañado, impidiendo la reparación (50, 52-55).
Estudios realizados en ratones transgénicos para HBx demostraron la aparición de la transversión G:CT:A en el codón 249 de TP53 como consecuencia de la exposición a AFB1; estos hallazgos demuestran que HBx aumenta la susceptibilidad de las células a la inducción de mutaciones inducidas por AFB1 (56, 57).
Estos resultados concuerdan con lo observado en los estudios epidemiológicos, en los cuales la mayor frecuencia de la mutación 249ser de TP53 se presenta en poblaciones con exposición crónica a AFB1 y con infección crónica por VHB. En particular el estudio prospectivo realizado por Kirk y otros autores en una población de Gambia aportó evidencia contundente del efecto multiplicativo de la infección crónica por VHB y el efecto mutagénico de AFB1 en TP53 (249ser) (58).
El riesgo de desarrollar CHC fue evaluado en 216 casos incidentes de CHC y 121 casos de cirrosis; este análisis demostró que el riesgo de desarrollar este tumor primario de hígado fue de 10 (razón de disparidad OR, 95%; IC: 5,16-19,6) en pacientes con marcador para la infección por VHB, de 13,2 (OR 95%; IC: 4,99-35,0) en pacientes con la mutación 249ser en TP53, mientras en pacientes con los dos marcadores el riesgo fue de 300 (OR 95%; IC: 48,6-3270).
Polimorfismos en XRCC1
El gen que codifica para la proteína x-ray repair cross-complementing group I (xrcc1) fue el primer gen aislado de mamífero involucrado en la sensibilidad a la radiación ionizante. A pesar de que ninguna actividad enzimática ha sido atribuida a esta proteína, se ha demostrado la asociación con la ADN polimerasa P (pol P), la ADN ligasa III (lig III) y la polimerasa poli-ADP-ribosa (PARP) para formar un complejo implicado en los mecanismos de reparación por escisión de bases (BER) y de reparación de quiebres de cadena sencilla (SSBR) (59). En el gen XRCC1 se han identificado tres polimorfismos en regiones codificantes: 194 ArgininaTriptofano (ArgTrp), 280 ArgininaHistidina (ArgHis) y 399 ArgininaGlutamina (ArgGln), siendo el polimorfismo 399Gln el más importante ya que está ubicado en el dominio de unión a PARP (60).
En diversos estudios se ha explorado la relación entre el polimorfismo 399Gln, la presencia de aductos de aflatoxina y el riesgo de desarrollar CHC. Lunn y otros autores evaluaron la relación entre los polimorfismos en XRCC1 y los niveles de aductos AFB1-ADN, en muestras de placenta provenientes de una población asiática con alta exposición a aflatoxina. Los individuos que presentaban el polimorfismo 399Gln tenían niveles superiores de aductos AFB1-ADN, comparados con los niveles observados en individuos con los polimorfismos 194Trp y 280His, lo cual sugiere que la xrcc1 participa en la reparación de los aductos AFB1-ADN (61). Otros estudios han confirmado que este polimorfismo está asociado con niveles elevados de aductos AFB1-ADN y mayor riesgo de desarrollar CHC (27, 31, 32).
Mutaciones en TP53
El rango de frecuencia de mutaciones de TP53 es de 5-50% dependiendo del tipo de tumor y del estadio (62). Las mutaciones por transición de nucleótidos predominan en cáncer de colon, cerebro y linfoblastomas, mientras mutaciones en regiones ricas en adenina y timina se encuentran asociadas con cáncer de esófago. Las mutaciones por transversión de nucleótidos, especialmente G:CT:A son más frecuentes en cáncer de hígado y cáncer de pulmón y ocurren en islas CpG (63). Mutaciones en el gen TP53 pueden modificar la vida media y la estabilidad de la proteína p53, su capacidad de unirse al ADN y la función transcripcional.
En CHC se han descrito mutaciones en TP53 en una proporción importante de los casos (9-11). Más de 80% de estas mutaciones corresponden a "missense mutations", es decir, producen cambios en la secuencia de aminoácidos que en la gran mayoría de casos generan una proteína no funcional (64).
Como se ha mencionado, en CHC se ha descrito una alta frecuencia de la mutación G:CT:A en la tercera posición del codón 249 en el exón 7 del gen TP53. Un 69,9% de las mutaciones reportadas en el codón 249 de TP53 que corresponden a la transversión G:CT:A han sido detectadas en CHC (64).
Esta mutación origina el cambio del aminoácido arginina por serina (mutación 249ser), alterando el dominio de unión de la proteína p53 al ADN. La presencia de esta mutación ha sido utilizada como marcador de exposición a AFB1 (8, 65, 66).
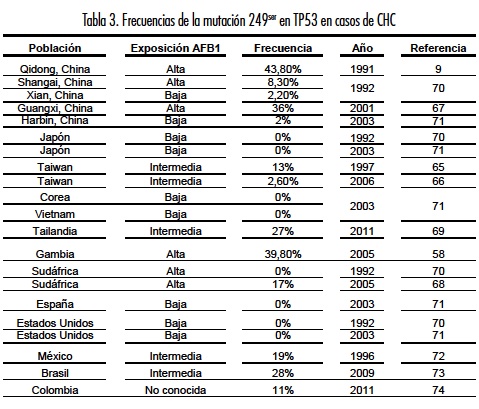
En diversas regiones del mundo se han realizado estudios para determinar la frecuencia de la mutación en casos de CHC. Estudios de poblaciones con exposición dietaría a aflatoxinas demostraron una alta frecuencia de la mutación en pacientes de las provincias de Qidong (43,8%) y Guangxi en China (36%) y en pacientes de Gambia (39,8%) (9, 67, 58). Por otra parte, en estudios realizados en regiones con exposición intermedia a aflatoxinas como Taiwán, Suráfrica y Tailandia, la mutación se ha demostrado en 13, 17 y 24% de los casos respectivamente (65, 68, 69). En países de mínima exposición a AFB1 como Estados Unidos, Japón, Corea, Vietnam y España la mutación ha sido detectada en menos de 5% de los casos de CHC analizados (70, 71).
En América Latina solo se han realizado dos trabajos encaminados a detectar la mutación 249ser en casos de CHC. El primer estudio corresponde a 16 casos de CHC en pacientes de México en los cuales se detectó la mutación 249ser en 19% de ellos (72). El segundo estudio fue realizado en Brasil con 74 casos de CHC; las muestras de tejido hepático fueron analizadas por dos técnicas, PCR y RFLP (polimorfismo de fragmentos de restricción) y por secuenciación directa. Un 28% de las muestras fue positivo para la mutación según la técnica de PCR y RFLP, mientras 16% de las muestras fue positiva según la técnica de secuenciación directa (73).
Aunque en Colombia existía evidencia de la presencia de aflatoxina en niveles superiores a los permitidos en muestras de maíz y arroz obtenidos en supermercados y plazas de mercado, la frecuencia de la mutación 249ser en la población no había sido explorada. Recientemente se publicó el primer estudio en pacientes con CHC en el que se evaluaron biomarcadores de exposición a AFB1, infección por VHB e infección por virus de la hepatitis C en muestras provenientes de pacientes con diagnóstico de CHC atendidos en cuatro centros hospitalarios de cuarto nivel de complejidad en las ciudades de Bogotá, Medellín y Cali. La exposición a la AFB1 se evaluó por detección de aductos utilizando un anticuerpo específico y por detección de la mutación por RFLP y secuenciación directa. El aducto no fue detectado en ninguna de las muestras, mientras que la mutación fue demostrada por las dos técnicas en 10,5% de los casos analizados (tabla 3) (74). Estos resultados (10,5%) son comparables con lo observado en el estudio en Brasil en el caso de las muestras analizadas por secuenciación directa (16%) y sugieren una exposición entre intermedia y baja en Colombia (74). Estudios adicionales son necesarios para confirmar lo descrito en este grupo de pacientes.
Conclusiones
La contaminación de alimentos para consumo humano con micotoxinas y el constante movimiento de los productos agrícolas alrededor del mundo plantean un problema de salud pública teniendo en cuenta el riesgo de exposición dietaría a este carcinógeno. Este riesgo es de particular interés en pacientes con infección crónica por VHB por el sinergismo demostrado entre estos dos factores de riesgo asociados al desarrollo de CHC; este carcinoma representa más de 80% de los casos de cáncer primario de hígado en el mundo.
La activación de la AFB1 por enzimas microsomales hepáticas y la capacidad de los metabolitos de reaccionar con el ADN representan la principal característica de este potente hepatocarcinógeno. La identificación de polimorfismos en genes que codifican enzimas que participan en la activación y detoxificación de la AFB1 puede considerarse como una variable importante para explorar en población con factores de riesgo para CHC.
Por otro lado, la detección de la mutación 249ser en muestras de suero puede ser utilizado como biomarcador en población general y en particular en pacientes con infección crónica por VHB, con el fin de identificar los individuos expuestos a otros factores de riesgo de CHC, además del VHB. Sin embargo, aún es necesario esclarecer en qué contexto puede ser aplicable este biomarcador, en detección temprana del CHC o simplemente como marcador de exposición a AFB1.
Las evidencias discutidas en esta revisión confirman la importancia de este hepatocarcinógeno y plantean la necesidad de monitorear la exposición de la población a AFB1.
Agradecimientos
Loa autores agradecen la financiación del Programa de Sostenibilidad de la Vicerrectoría de Investigación de la Universidad de Antioquia.
Descargos de responsabilidad
Los autores están de acuerdo con el contenido del artículo y son responsables del mismo. Declaran no tener conflicto de intereses.
Bibliografía
1. Smela ME, Currier SS, Bailey EA, Essigmann JM. The chemistry and biology of aflatoxin B(1): from mutational spectrometry to carcinogenesis. Carcinogenesis 2001; 22 (4): 535-45. [ Links ]
2. Moss MO. Mycotoxins. Mycol Res 1996; 100 (5): 513-23. [ Links ]
3. Jelinek CF, Pohland AE, Wood GE. Worldwide ocurrence of mycotoxins in foods and feeds and update. J Assoc Off Anal Chem 1989; 72 (2): 223-30. [ Links ]
4. International Agency for Research on Cancer. Monographs on the evaluation of carcinogenic risks to humans, Vol. 82. Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. Lyon: IARC; 2002. [ Links ]
5. Williams JH, Phillips TD, Jolly PE, Stiles JK, Jolly CM, Aggarwal D. Human aflatoxicosis in developing countries: a review of toxicology, exposure, potential health consequences, and interventions. Am J Clin Nutr 2004; 80 (5): 1106-22. [ Links ]
6. Bennett JW, Klich M. Mycotoxins. Clin Microbiol Rev 2003; 16 (3): 497-516. [ Links ]
7. International Agency for Research on Cancer. The evaluation of the carcinogenic risk of chemicals to humans. IARC Monograph Supplement 4. Lyon: IARC; 1982. [ Links ]
8. Aguilar F, Hussain P, Cerutti P. Aflatoxin B1 induces the transversion G-T in codon 249 of the p53 tumor suppressor gene in human hepatocytes. PNAS 1993; 90 (18): 8586-90. [ Links ]
9. Ozturk M. P53 mutation in hepatocellular carcinoma after aflatoxin exposure. Lancet 1991; 338 (8779): 1356-9. [ Links ]
10. Bressac B, Kew M, Wands J, Ozturk M. Selective G to T mutations of p53 gene in Hepatocellular carcinoma from Southern Africa. Nature 1991; 350 (6317): 429-31. [ Links ]
11. Smela ME, Hamm ML, Henderson PT, Harris CM, Harris TM, Essigmann JM. The aflatoxin B(1) formamidopyrimidine adduct plays a major role in causing the types of mutations observed in human hepatocellular carcinoma. PNAS 2002; 99 (10): 6655-60. [ Links ]
12. Szymanska K, Lesi OA, Kirk GD, Sam O, Taniere P, Scoazec JY et al. Ser-249TP53 mutation in tumour and plasma DNA of hepatocellular carcinoma patients from a high incidence area in the Gambia, West Africa. Int J Cancer 2004; 110 (3): 374-9. [ Links ]
13. De Moura Gallo CV, Azevedo E Silva MG, de Moraes E, Olivier M, Hainaut P. TP53 mutations as biomarkers for cancer epidemiology in Latin America: current knowledge and perspectives. Mutat Res 2005; 589 (3): 192-207. [ Links ]
14. Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. Mutational hotspot in the p53 gene in human hepatocellular carcinoma. Nature 1991; 350 (6317): 427-8. [ Links ]
15. Hayward NK, Walker GJ, Graham W, Cooksley E. Hepatocellular carcinoma mutation. Nature 1991; 352 (6338): 764. [ Links ]
16. Okuda K. Hepatocellular Carcinoma-history current status and perspectives. Dig Liver Dis 2002; 34 (9): 613-6. [ Links ]
17. Forrester LM, Neal GE, Judah DJ, Glancey MJ, Wolf RC. Evidence for involvement of multiple forms of cytochrome P-450 in aflatoxin B1 metabolism in human liver. PNAS 1990; 87 (21): 8306-10. [ Links ]
18. Gallagher EP, Kunze KL, Stapleton PL, Eaton DL. The kinetics of aflatoxin B1 oxidation by human cDNA-expressed and human liver microsomal cytochromes P450 1A2 and 3A4. Toxicol Appl Pharmacol 1996; 141 (2): 595-606. [ Links ]
19. Wang H, Dick R, Yin H, Licad-Coles E, Kroetz DL, Szklarz G et al. Structure-function relationships of human liver cytochromes P450 3A: aflatoxin B1 metabolism as a probe. Biochemistry 1998; 37 (36): 12536-45. [ Links ]
20. Chen X, Wang H, Xie W, Liang R, Wei Z, Zhi L et al. Association of CYP1A2 genetic polymorphisms with hepatocellular carcinoma susceptibility: a case-control study in a high-risk region of China. Pharmacogenet Genomics 2006; 16 (3): 219-27. [ Links ]
21. Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet 2001; 27 (4): 383-91 [ Links ]
22. Hustert E, Haberl M, Burk O, Wolbold R, He YQ, Klein K et al. The genetic determinants of the CYP3A5 polymorphism. Pharmacogenetics 2001; 11 (9): 773-9. [ Links ]
23. Wojnowski L, Turner PC, Pedersen B, Hustert E, Brockmoller J, Mendye M et al. Increased levels of aflatoxin-albumin adducts are associated with CYP3A5 polymorphisms in The Gambia, West Africa. Pharmacogenetics 2004; 14 (10): 691-700. [ Links ]
24. Guengerich FP, Johnson WW, Shimada T, Ueng YF yamazaki H, Langouet S. Activation and detoxication of aflatoxin B1. Mutat Res 1998; 402 (1-2): 121-8. [ Links ]
25. Kelly EJ, Erickson KE, Sengstag C, Eaton DL. Expression of Human microsomal epoxide hydrolase in Saccharomyces cerevisiae reveals a functional role in aflatoxin B1 detoxification. Toxicol Sci 2002; 65 (1): 35-42. [ Links ]
26. Sun CA, Wang LY, Chen CJ, Lu SN, You SL, Wang LW et al. Genetic polymorphism of glutathione S-transferases M1 and T1 associated with susceptibility-related hepatocarcinogenesis among chronic hepatitis B carriers: a nested case-control study in Taiwan. Carcinogenesis 2001; 22 (8): 1289-94. [ Links ]
27. Kirk GD, Turner PC, Gong Y, Lesi OA, Mendy M, Goedert JJ et al. hepatocellular carcinoma and polymorphisms in carcinogen-metabolizing and dna repair enzymes in a population with aflatoxin exposure and hepatitis B virus endemicity. Cancer Epidemiol Biomarkers Prev 2005; 14 (2): 373-9. [ Links ]
28. McGlynn KA, Rosvold EA, Lustbader ED, Hu Y, Clapper ML, Zhou T et al. Susceptibility to hepatocellular carcinoma is associated with genetic variation in the enzymatic detoxification of aflatoxin B1. PNAS 1995; 92 (6): 2384-7. [ Links ]
29. Omer RE, Verhoef L, Veer PV, Idris MO, Kadaru AMY, Kampman E et al. Peanut butter intake, GSTM1 genotype and hepatocellular carcinoma: a case-control study in Sudan. Cancer Causes Control 2001; 12 (1): 23-32. [ Links ]
30. Tiemersma EW, Omer RE, Bunschoten A, Veer PV, Kok FJ, Idris MO et al. Role of genetic polymorphism of glutathione-s-transferase t1 and microsomal epoxide hydrolase in aflatoxin-associated hepatocellular carcinoma. Cancer Epidemiol Biomarkers Prev 2001; 10 (7): 785-9. [ Links ]
31. Yu MW yang SY, Pan IJ, Lin CL, Liu CJ, Liaw YF et al. Polymorphisms in XRCC1 and glutathione S-Transferase Genes and Hepatitis B-related hepatocellular carcinoma. J Natl Cancer Inst 2003; 95 (19): 1485-8. [ Links ]
32. Long XD, Ma Y, Wei YP, Deng ZL. The polymorphisms of GSTM1, GSTT1, HYL1*2, and XRCC1, and aflatoxin B1-related hepatocellular carcinoma in Guangxi population, China. Hepatol Res 2006; 36 (1): 48-55. [ Links ]
33. Hassett C, Aicher L, Sidhu JS, Omiecinski CJ. Human microsomal epoxide hydrolase: genetic polymorphism and functional expression in vitro of amino acid variants. Human Mol Genet 1994; 3 (3): 421-8. [ Links ]
34. McGlynn KA, Hunter K, LeVoyer T, Roush J, Wise P, Michielli RA et al. Susceptibility to aflatoxin B1-related primary hepatocellular carcinoma in mice and humans. Cancer Res 2003; 63 (15): 4594-601. [ Links ]
35. Kobertz WR, Wang D, Wogan GN, Essigmann JM. An intercalation inhibitor altering the target selectivity of DNA damaging agents: Synthesis of site-specific aflatoxin B1 adducts in a p53 mutational hotspot. PNAS 1997; 94 (18): 9579-84. [ Links ]
36. Benasutti M, Ejadi S, Whitlow MD, Loechler EL. Mapping the binding site of aflatoxin B1 in DNA: Systematic analysis of the reactivitybof aflatoxina B1 with guanines in different DNA sequences. Biochemistry 1988; 27 (1): 472-81. [ Links ]
37. Denissenko MF, Cahill J, Koudriakova TB, Gerber N, Pfeifer GP. Quantitation and mapping of aflatoxin B1-induced DNA damage in genomic DNA using aflatoxin B1-8,9-epoxide and microsomal activation systems. Mutat Res 1999; 425 (2): 205-11. [ Links ]
38. Jones WR, Johnston DS, Stone MP. Site-Specific Synthesis of Aflatoxin B1 Adducts within an oligodeoxyribonucleotide containing the human p53 codon 249 sequence. Chem Res Toxicol 1999; 12 (8): 707-14. [ Links ]
39. Denissenko MF, Chen JX, Tang MS, Pfeifer GP. Cytosine methylation determines hot spots of DNA damage in the human P53 gene. PNAS 1997; 94 (8): 3893-8. [ Links ]
40. Chen JX, Zheng Y, West M, Tang MS. Carcinogens preferentially bind at methylated CpG in the p53 mutational hot spots. Cancer Res 1998; 58 (10): 2070-5. [ Links ]
41. Hoeijmakers JH. Genome maintenance mechanism for preventing cancer. Nature 2001; 411 (6835): 366-74. [ Links ]
42. Bedard LL, Massey TE. Aflatoxin B1-induced DNA damage and its repair. Cancer Lett 2006; 241 (2): 174-83. [ Links ]
43. Alekseyev YO, Hamm ML, Essigmann JM. Aflatoxin B1 formamidopyrimidine adducts are preferentially repaired by the nucleotide excision repair pathway in vivo. Carcinogenesis 2004; 25 (6): 1045-51. [ Links ]
44. Guo Y, Breeden LL, Zarbl H, Preston BD, Eaton DL. Expression of a human cytochrome P450 in yeast permits analysis of pathways for response to and repair of aflatoxin-induced DNA damage. Mol Cell Biol 2005; 25 (14): 5823-33. [ Links ]
45. Leadon SA, Tyrrell RM, Cerutti PA. Excision repair of aflatoxin B1-DNA adducts in human fibroblasts. Cancer Res 1981; 41 (12): 5125-9. [ Links ]
46. Levy DD, Groopman JD, Lim SE, Seidman MM, Kraemer KH. Sequence specificity of aflatoxin B1-induced mutations in a plasmid replicated in xeroderma pigmentosum and DNA repair proficient human cells. Cancer Res 1992; 52 (20): 5668-73. [ Links ]
47. Takahashi Y, Nakatsuru Y, Zhang S, Shimizu Y, Kume H, Tanaka K et al. Enhanced spontaneous and aflatoxin-induced liver tumorigenesis in xeroderma pigmentosum group A gene deficient mice. Carcinogenesis 2002; 23 (4): 627-33. [ Links ]
48. Qian GS, Ross RK yu MC. A follow-up study of urinary markers of aflatoxin exposure and liver cancer risk in Shanghai, People's Republic of China. Cancer Epidemiol Biomarkers Prev 1994; 3 (1): 3-10. [ Links ]
49. Ming L, Thorgeirsson SS, Gail MH, Lu P, Harris CC, Wang N et al. Dominant role of hepatitis B virus and cofactor role of aflatoxin in hepatocarcinogenesis in Qidong, China. Hepatology 2002; 36 (5): 1214-20. [ Links ]
50. Wang XW, Forrester K, Yeh H, Feitelson M.A, Gu J, Harris C.C. Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding, transcriptional activity and association with ERCC3. PNAS 1994; 91 (6): 2230-4. [ Links ]
51. Prost S, Ford C, Taylor C, Doig J, Harrison DJ. Hepatitis B x protein inhibitis p53 dependent DNA repair in primary mouse hepatocytes. J Biol Chem 1998; 273 (50): 33327-32. [ Links ]
52. Qadri I, Conaway JW, Conaway RC Schaack J, Siddiqui A. Hepatitis B virus transactivator protein, HBx, associates with the components of TFIIH and stimulates the DNA helicase activity of TFIIH. PNAS 1996; 93 (20): 10578-83. [ Links ]
53. Jia L, Wang XW, Harris CC. Hepatitis B virus X protein inhibits nucleotide excision repair. Int J Cancer 1999; 80 (6): 875-9. [ Links ]
54. Groisman IJ, Benlimame N, Slagle BL, Perez MH, Alpert L, Song DJ et al. Transcriptional regulation of the TFIIH transcription repair components XPB and XPD by the hepatitis B virus x protein in liver cells and transgenic liver tissue. J Biol Chem 2001; 276 (17): 14124-32. [ Links ]
55. Capovilla A, Carmona S, Arbuthnot P. Hepatits B virus X protein binds damage DNA and sensitizes liver cells to ultraviolet irradiation. Biochem Biophys Res Commun 1997; 232 (1): 255-60. [ Links ]
56. Sohn S, Jaitovitch-Groisman I, Benlimame N, Galipeau J, Batist G, Alaoui-Jamali MA. Retroviral expression of the hepatitis B virus x gene promotes liver cell susceptibility to carcinogen-induced site specific mutagenesis. Mutat Res 2000; 460 (1): 17-28. [ Links ]
57. Madden CR, Finegold MJ, Slagle BL. Altered DNA mutation spectrum in aflatoxin B1-treated transgenic mice that express the hepatitis B virus X protein. J Virol 2002; 76 (22): 11770-4. [ Links ]
58. Kirk GD, Lesi OA, Mendy M, Szymanska K, Whittle H, Goedert JJ et al. 249ser TP53 mutation in plasma DNA, hepatitis B viral infection, and risk of hepatocellular carcinoma. Oncogene 2005; 24 (38): 5858-67. [ Links ]
59. Thompson LH & West MG. XRCC1 keeps DNA from getting stranded. Mutat Res 2000; 459 (1): 1-18. [ Links ]
60. Shen MR, Jones IM, Mohrenweiser H. Nonconservative Amino acid substitution variants exist at polymorphic frequency in DNA repair genes in healthy humans. Cancer Res 1998; 58 (4): 604-8. [ Links ]
61. Lunn RM, Langlois RG, Hsieh LL, Thompson CL, Bell DA. XRCC1 polymorphisms: effects on aflatoxin B1-DNA adducts and glycophorin a variant frequency. Cancer Res 1999; 59 (11): 2557-61. [ Links ]
62. Hussain SP, Schwank J, Staib F, Wang XW, Harris CC. TP53 mutations and hepatocellular carcinoma: insights into the etiology and pathogenesis of liver cancer. Oncogene 2007; 26 (15): 2166-76. [ Links ]
63. Hollstein M, Sidransky D, Volgenstein B, Harris CC. P53 mutations in human cancers. Science 1991; 5 (253): 49-53. [ Links ]
64. International Agency for Research on Cancer. Lyon: The International Agency for Research on Cancer; 2006. IARC TP53 Mutation Database. [Consultado el 21 de noviembre de 2011]. Disponible en http://www-p53.iarc.fr/ [ Links ]
65. Lunn RM, Zhang YJ, Wang LY, Chen CJ, Lee PH, Lee CS et al. P53 mutations, chronic hepatitis B virus infection, and aflatoxin exposure in hepatocellular carcinoma in Taiwan. Cancer Res 1997; 57 (16): 3471-7. [ Links ]
66. Zhang YJ, Rossner Jr P, Chen Y, Agrawal M, Wang Q, Wang L et al. Aflatoxin B1 and polycyclic aromatic hydrocarbon adducts, p53 mutations and p16 methylation in liver tissue and plasma of hepatocellular carcinoma patients. Int J Cancer 2006; 119 (5): 985-91. [ Links ]
67. Stern MC, Umbach DM yu MC, London SJ, Zhang ZQ, Taylor JA. Hepatitis B, aflatoxin B1, and p53 codon 249 mutation in hepatocellular carcinomas from Guangxi People's Republic of China, and a meta-analysis of existing studies. Cancer Epidemiol Biomarkers Prev 2001; 10 (6): 617-25. [ Links ]
68. Kimbi GC, Kew MC, Yu MC, Arakawa K, Hodkinson J. 249ser p53 mutation in the serum of black southern African patients with hepatocelular carcinoma. J Gastroenterol Hepatol 2005; 20 (8): 1185-90. [ Links ]
69. Galy O, Chemin I, Le Roux E, Villar S, Le Calvez-Kelm F, Lereau M et al. Mutations in TP53 and CTNNB1 in relation to hepatitis B and C infections in hepatocellular carcinomas from Thailand. Hepat Res Treat 2011; 2011 (2011): 1-9. [ Links ]
70. Buetow KH, Sheffield VA, Zhu M, Zhou T, Shen FM, Hino O et al. Low frequency of p53 mutations observed in a diverse collection of primary hepatocellular carcinomas. PNAS 1992; 89 (20): 9622-6. [ Links ]
71. Ding Xin, Park NJ, Taltavull TC, Thung SN, Jin X, Jin Y et al. Geographic characterization of hepatitis virus infections, genotyping of hepatitis B virus and p53 mutation in hepatocellular carcinoma analized by in situ detection of viral genome from carcinoma tissues: comparison among six different countries. Jpn J Infect Dis 2003; 56 (1): 12-8. [ Links ]
72. Soini Y, Chia SC, Bennett WP, Groopman JD, Wang JS, Benedetti VMG et al. An aflatoxin-associated mutational hotspot at codon 249 in the p53 tumor suppressor gene occurs in hepatocellular carcinomas from Mexico. Carcinogenesis 1996; 17 (5): 1007-12. [ Links ]
73. Nogueira JA, Ono-Nita SK, Nita ME, de Souza MM, do Carmo EP, Mello ES et al. 249 TP53 mutation has high prevalence and is correlated with larger and poorly differentiated HCC in Brazilian patients. BMC Cancer 2009; 9 (204): 1-8. [ Links ]
74. Navas MC, Suarez I, Carreño-Venegas A, Uribe D, Ríos WA, Cortés-Mancera F et al. Hepatitis B and Hepatitis C infection biomarkers and TP53 mutations in hepatocellular carcinomas from Colombia. Hepat Res Treat 2011; 2011 (2011): 1-10. [ Links ]
