











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink1. INTRODUCCIÓN
Una estructura cristalina puede considerarse como un conjunto de capas o planos, los cuales actúan de forma similar a un espejo semitransparente. Los rayos X que tienen una longitud de onda proporcional a la distancia interplanar se pueden reflejar de tal forma que cumplen la ley de reflexión. A este comportamiento se le conoce como difracción y es descrito por la ley de Bragg. Cuando ocurre una interferencia constructiva debido a los haces difractados de rayos X, esta ley se cumple, y en este caso se tiene una reflexión de Bragg, la cual se recoge mediante un detector de barrido.
La difracción de rayos X (DRX) es una técnica utilizada para la caracterización estructural de materiales cristalinos. Esta técnica permite determinar la estructura cristalina de un material, al igual que las fases y las imperfecciones que este pueda tener.
El físico holandés Rietveld, desarrolló el método de Rietveld, siendo este una técnica de refinamiento que consiste en minimizar la diferencia entre un modelo cristalográfico teórico y un patrón de difracción experimental, haciendo uso del método de mínimos cuadrados 1. El modelado de altura, anchura, intensidad y posición de las reflexiones de Bragg en un patrón de difracción de rayos X, puede producir una gran cantidad de información acerca de la estructura cristalina de un material, tales como los parámetros de red, posiciones atómicas, tamaño del cristal, distancias interplanares, cuantificación de las fases cristalinas de la muestra y microdeformaciones, entre otras.
Dentro de los programas que están disponibles en forma libre para desarrollar refinamiento de estructuras se encuentran GSAS, Fullprof, DBWS.
El tantalato de itrio y niobato de itrio, también conocidos como estructuras fergusonitas, son importantes fósforos, siendo estos materiales emisores de luz visible como resultado de la absorción de energía 2. Cuando la energía absorbida es procedente de los rayos X, estos reciben el nombre de fósforos de rayos X, los cuales son materiales inorgánicos de estado sólido que se usan en aplicaciones médicas de imágenes diagnósticas, siendo su función la de reducir la exposición del paciente sin alterar las características de la imagen.
Los fósforos han sido ampliamente estudiados desde la década de los 70 3-6. En estos trabajos se encontró mediante refinación que el tantalato de itrio tiene 3 estructuras cristalinas, una tetragonal (estructura T) a altas temperaturas (>1450 °C), una monoclínica (estructura M) a bajas temperaturas (<1400 °C), y otra estructura monoclínica M' que puede ser sintetizada a 1400°C. Generalmente, la estructura M' se utiliza como material luminiscente. Para la estructura M'- YTaO4 fueron encontrados los siguientes parámetros cristalográficos: a=5.298 Å, β =5.451 Å, =5.111 Å, p=96.45° y una densidad de 7.56 g/cm3. A temperaturas superiores a 1450 °C, M'-YTaO4 se convierte en una estructura T (esquelita I44/a) y al enfriarse en M (fergusonita). Los átomos de tantalio se encuentran en una coordinación octaédrica con seis enlaces de Ta O. Los átomos de itrio están rodeados por 8 átomos de oxígeno formando un cubo distorsionado. Dentro de la estructura M'-YTaO4 se encuentran 2 átomos de itrio, 2 de tantalio y 8 de oxígeno.
El niobato de itrio (YNbO4) presenta dos tipos de estructura, una estructura tetragonal formada a altas temperaturas (T-esquelita I44/a) y una estructura M formada a bajas temperaturas con grupo espacial I2-C2 3 y simetría puntual C2 para el grupo molecular NbO4. La transición entre las dos fases ocurre de forma reversible a los 800 °C. Los átomos de Nb están en coordinación octaédrica con seis enlaces Nb-O, de igual forma que la fergusonita M'-YTaO4. El volumen de la celda M-YNbO4 es el doble de M'-YTaO4. En esta estructura se encuentran 4 átomos de itrio, 4 de niobio y 16 de oxígeno 7-11.
Actualmente, se está estudiando la aplicación de estos fósforos en pantallas de emisión de campo (FED), diodos emisores de luz UV y pantallas intensificadoras de rayos X 12-18.
2. MARCO TEÓRICO
El método de Rietveld permite refinar una estructura cristalina mediante un ajuste basado en modelos teóricos y experimentales, usando el método de mínimos cuadrados. El principio del método de Rietveld consiste en minimizar una función M, la cual analiza la diferencia entre una intensidad observada y una calculada. Rietveld definió esta función como función residuo, y se calcula mediante la ecuación 1:
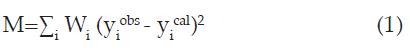
Donde W es el peso asignado a cada punto y se determina experimentalmente a partir de Wi = 1/(Iexp (20i)), y.obs es la intensidad observada experimental en el punto i-ésimo del difractograma, es la intensidad calculada en el punto i-ésimo, mientras que la sumatoria se realiza sobre todos los puntos del patrón de difracción.
La función residuo es una función compleja que incluye todos los parámetros que dan lugar a un patrón de difracción. El refinamiento consiste en encontrar los valores óptimos de estos parámetros de manera que M adquiera el valor mínimo posible 19.
Se requieren dos tipos de datos para obtener las intensidades calculadas. Uno de ellos son los llamados parámetros estructurales o atómicos (parámetros de red, grupo espacial, posiciones atómicas, factores de agitación térmicos) y el otro tipo de datos son los parámetros globales, dentro de los cuales se encuentran: factor de escala del difractograma, desplazamiento del cero, parámetros que describen la forma de los picos, parámetros que corrigen la absorción, etc.
2.1 Cálculo de la intensidad de los picos de difracción
La intensidad calculada de un pico de difracción, yi cal, para cada punto 20i se determina mediante la ecuación (2):
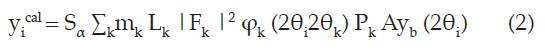
Siendo Sa es el factor de escala, que depende de la muestra irradiada, representa los índices de Miller (h k l) para una reflexión de Bragg, mk es la multiplicidad de la reflexión, L, contiene los factores de corrección de Lorentz y polarización, |Fk |2 es el factor estructural para la reflexión k-ésima de Bragg, ϕk (20i - 20k) es una función que describe la forma de la reflexión, Pk es una función que puede corregir los efectos de orientación del cristal, A es un factor de corrección de absorción que se relaciona con el grosor de la muestra y de la geometría del difractómetro, yb (20i) es la contribución del fondo del difractograma en el punto 20i19.
2.2 Cálculo del factor estructural
El factor estructural indica la capacidad de difracción de todos los átomos de una celda unitaria, este factor se calcula a través de la ecuación (3):
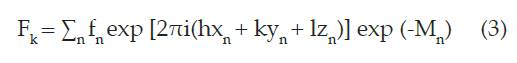
En esta ecuación, fn es el factor atómico de dispersión del n-ésimo átomo, h, k, l son los índices de Miller que producen la difracción, xn, yn, zn son las posiciones de los átomos en la celda unitaria, Mn es Bj sin2 θ/ λ2, donde Bj es el coeficiente de temperatura isotrópico y está relacionado con la vibración atómica debido a la temperatura y θ es el ángulo de incidencia del haz de rayos X 20.
Como ya se indicó, los parámetros que se refinan en el método Rietveld son los estructurales y los globales; los primeros hacen parte del factor estructural, y los segundos hacen parte de la ecuación (2) y afectan todo el difractograma.
2.3. Funciones usadas para el modelamiento del perfil del pico de difracción
La descripción de la forma de los picos de difracción es un proceso demasiado importante en el método de Rietveld. Además de una función perfil que modele los picos de difracción, son necesarias funciones que modelen la anchura a media altura (FWHM), la asimetría y el fondo. Las funciones utilizadas para modelar el pico de difracción son descritas por tres parámetros, posición (20k), intensidad (I0) y el ancho a media altura (FWHM) 21. Algunas de estas funciones son:
Gaussiana (G)
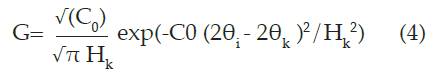
Donde Cn = 4ln2, Hk, es el ancho de la altura media del pico de difracción para la k-ésima reflexión y (20i -20k) es el ángulo de Bragg para la k-ésima reflexión.
Lorentzianna (L)
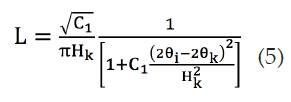
Donde y los demás valores son los ya definidos para la función Gaussiana
Pseudo - Voigt (pV)
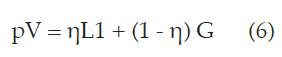
La relación existente entre el ensanchamiento total del pico de difracción y los ensanchamientos parciales de los picos de difracción de la componente Gaussiana y Lorentziana ( 28 ) , dados por las ecuaciones (7), (8) y (9):
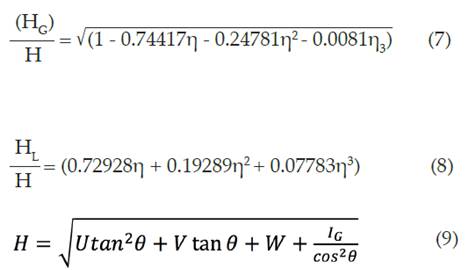
donde H es el ensanchamiento total del pico de difracción y se obtiene a través del refinamiento del ensanchamiento debido a factores instrumentales (U, V, W) e IG (ensanchamiento por tamaño de cristalitos). La ecuación (9) se basa en la teoría de Caglioti 23.
Usualmente el perfil del pico de difracción es ajustado a este tipo de función, ya que contiene tanto la función Gaussiana como Lorentziana. Esta última describe mejor el ensanchamiento del pico de difracción debido al tamaño de los cristalitos de la muestra de polvo, mientras que la función Gaussiana representa mejor la forma del pico debido a factores instrumentales. Si la forma del pico de difracción se ajusta a la Gaussiana, el parámetro q=0, pero, por otro lado, si tiene forma Lorentziana, entonces q=1. Se debe considerar la situación donde el pico de difracción tiene una combinación de ambas funciones, para la cual el valor de q estará entre 0 y 1, dependiendo de la tendencia de la función siendo 0.75 el más usado.
2.4. Ajuste del fondo de un difractograma
Cuando se tiene una muestra puramente cristalina (sin defectos ni fases amorfas), el difractograma se puede modelar por convolución de las intensidades de Bragg, calculadas según el equipo de difracción y la contribución de fondo debida a la dispersión del aire y a los factores térmicos 24. Para realizar el ajuste del fondo se pueden hacer dos cosas: realizar una estimación por interpolación lineal entre puntos seleccionados situados en el pico de difracción o utilizar una función que modele el fondo. Ambos métodos funcionan bien cuando los picos de difracción están bien resueltos.
2.5. Validez de un ajuste para el refinamiento
En el método de Rietveld existen criterios que sirven para estimar la validez de un ajuste, los cuales ayudan a decidir si el modelo propuesto es el correcto para que el usuario pueda determinar cuándo detener el refinamiento. Los criterios más usados son:
Residuo del patrón pesado (Rwp): este criterio muestra el progreso del refinamiento y se calcula mediante la ecuación (10):
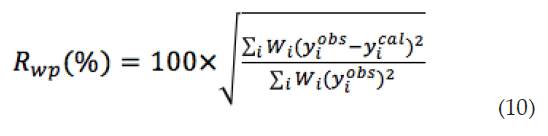
La definición de las diferentes variables ya se ha comentado a lo largo del artículo.
Residuo del peso (Rp): está relacionado con el Residuo del patrón pesado, y evalúa cada uno de los puntos del difractograma. Rp se calcula con la ecuación (11):
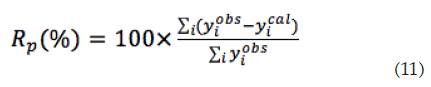
Residuo del factor de Bragg (R b ): indica la calidad de los datos cristalográficos de la celda unitaria (grupo espacial, parámetros de red, posición de los átomos y el número de ocupación de los átomos). La fórmula para calcular el residuo de Bragg se expresa través de la ecuación (12):
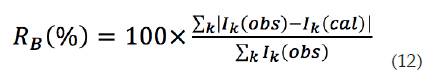
Factor estadístico esperado (Rexp): el valor final de debe aproximarse al factor estadístico esperado . Este criterio refleja la calidad de los datos obtenidos en la medición del patrón de difracción y se calcula por medio de la ecuación (13):
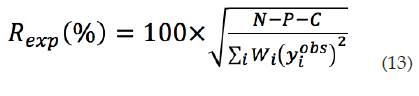
Donde N es elnúmero de observaciones independientes, P es el número de parámetros refinados y C, el número de ecuaciones que restringen el refinamiento.
Bondad del ajuste (X 2 ): es el cociente entre Rwp y Rexp

Para un caso ideal, el valor de X2 debe estar entre 1 y 1.3 Si el tiempo de toma de datos es grande y se tiene un tamaño de paso pequeño, los errores estadísticos no dominarán, R exp podría ser pequeño y X2 sería mayor que la unidad. Este es el caso habitual en los refinamientos de un cristal donde son habituales los valores entre 5 y 20. Por otro lado, si el tiempo de toma de datos es pequeño y el tamaño del paso es demasiado grande, R exp es grande y X2 es menor que 1.
Los criterios descritos anteriormente son importantes para la evaluación de un refinamiento, pero el más importante es el ajuste de los datos del patrón calculado con los datos del patrón observado, es decir, en el método de Rietveld se debe incluir la gráfica con las intensidades calculadas, las intensidades observadas y la diferencia entre éstas 24.
3. METODOLOGÍA
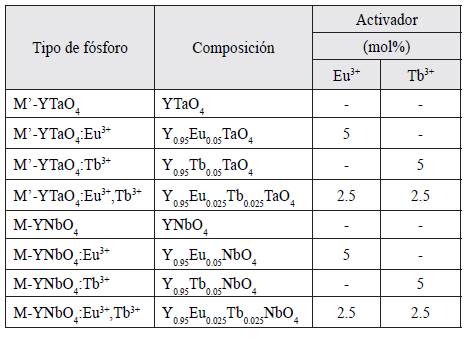
Las muestras de YTaO4 y YNbO4 dopadas con Eu3+ y Tb3+ fueron preparadas mediante el método de reacción de estado sólido, usando mezclas homogéneas de Y2O3, Ta2O5 y Nb2O5, y NaSO4 como fundente. Para el dopaje se utilizó Eu2O3 y Tb2O3. Las mezclas se homogeneizaron con acetona, y se secaron a 70°C. Después se hornearon a 1200°C durante 4 horas y se enfriaron lentamente hasta temperatura ambiente. Finalmente las muestras se lavaron con agua, se secaron y tamizaron. La composición química de los fósforos preparados se puede ver en la tabla 1.
Las propiedades estructurales fueron estudiadas mediante una fuente de radiación Cu-Kα (λ=1.5405 Å) generada por un ánodo rotatorio Rigaku, operando a 40 kV y 40 mA. La geometría usada fue Bragg-Brentano realizándose un barrido desde los 10° a 80° (20) con un paso de 0.02° para obtener los patrones de difracción. El análisis morfológico fue llevado a cabo con un microscopio electrónico de barrido Hitachi S-4700.
4. RESULTADOS
4.1 Análisis morfológico
En la figura 1 se ven las diferencias morfológicas entre las estructuras YTaO4 y YNbO4 tomadas con un microscopio electrónico de barrido. En las imágenes se puede apreciar una buena morfología, caracterizada por granos limpios, los cuales se pueden obtener usando fundentes basados en sulfatos (SO4). Los granos de la estructura YTaO4 (figura 1a) tienen forma poliédrica alargada, y un tamaño entre 2 y 5 μm.

Figura 1 Imágenes de las muestras a) YTaO4 y b) YNbO4 tomadas por un microscopio electrónico de barrido.
El tamaño de los granos de la estructura YNbO4 (figura 1b) está entre 10 y 12 μm, y tienen una forma poliédrica más redonda. Las dimensiones micrométricas de estos granos se encuentran en el rango deseable para el análisis por difracción de rayos X. Las dimensiones de los granos sintetizados son ideales para el estudio de propiedades luminiscentes de estos compuestos, investigadores han utilizado dopajes de hasta 30% Tb3+, obteniendo tamaño de granos similares al de nuestro trabajo 25. El tamaño ideal de los cristales debe estar entre 3 y 12 μm para tales aplicaciones como pantallas de emisión de campo (FED), diodos emisores de luz UV y pantallas intensificadoras de rayos X, ya que un tamaño de partículas más grandes causan problemas a la hora de su fabricación, y partículas más pequeñas pierden intensidad de emisión a través de dispersión interna 26.
Además, se usaron los parámetros Lx y Ptec del programa GSAS, estos parámetros permiten calcular el tamaño promedio del grano de forma perpendicular y longitudinal. En la estructura YTaO4 se encontró un tamaño perpendicular promedio de 2.2 μm y de 5.1 μm para el tamaño promedio longitudinal. En el caso del YNbO4, el tamaño perpendicular es de 6.7 μm y el longitudinal de 12.8 μm. Estos datos son congruentes con las dimensiones obtenidas en las imágenes SEM.
4.2 Caracterización estructural por difracción de rayos X
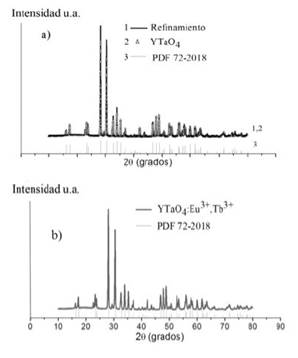
En las figuras 2 y 3se presentan superpuestos los patrones de difracción de rayos X de los compuestos estudiados, el refinamiento Rietveld (línea azul) y el patrón de difracción obtenido de la base de datos ICDD (International Center for Diffraction Data) (línea magenta).
En las figuras 2a) y 3a) las estructuras YTaO4 y YNbO4 están sin dopaje, mientras que a las estructuras de las figuras 2b) y 3b) se les realizó una sustitución de 5% de átomos de itrio por 2.5% de iones de Eu3+ y 2.5% de iones de Tb3+. Según la base de datos ICDD estos compuestos tienen una tarjeta PDF (Powder Diffraction File) 72-2018 y 72-2077, lo que evidencia que el fósforo YTaO4 presenta una estructura monoclínica M'-YTaO4, y el fósforo YNbO4 presenta una estructura monoclínica M-YNbO4. Si se comparan las partes a) y b) de las figuras 2 y 3 (líneas azules y verdes), se puede notar que todos los picos están en la misma posición y tienen las mismas intensidades, ambos patrones de difracción son exactamente iguales, lo que indica que la estructura no varía al introducir un defecto sustitucional de átomos de itrio (Y) por iones de Eu3+ y Tb3+ en las fergusonitas YTaO4 y YNbO4. Dicha sustitución conlleva a un incremento en el volumen de la celda unitaria de acuerdo con la ley de Vegard. Estos valores fueron calculados y se presentan en la tabla 2.
Para el compuesto tantalato de itrio, el defecto sustitucional creado aumentó el volumen de la celda original en 0.284% cuando se agregaron impurezas de Eu3+ (YTaO4:Eu3+). Cuando se utilizó iones de Tb3+ (YTaO4:Tb3+), el aumento en la celda unitaria fue de 0.151% y de 0.189% para la doble incorporación de iones de europio y terbio, (YTaO4:Eu3+,Tb3+) en red cristalina.
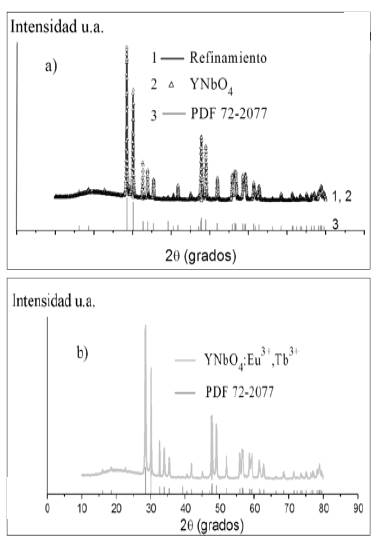
Tabla 2 Constantes de red y volumen de las celdas unitarias de los fósforos tantalato de itrio y niobato de itrio dopados con Eu3+ y/o Tb3+.
| Tipo de fósforo | a (Å) | b (Å) | c (Å) | β | V (Å 3) |
|---|---|---|---|---|---|
| YTaO4 | 5.293 | 5.456 | 5.107 | 96.36 | 146.575 |
| YTaO4:Eu3+ | 5.298 | 5.461 | 5.112 | 96.36 | 146.992 |
| YTaO4:Tb3+ | 5.296 | 5.459 | 5.109 | 96.36 | 146.796 |
| YTaO4:Eu3+,Tb3+ | 5.296 | 5.46 | 5.11 | 96.36 | 146.852 |
| YNbO4 | 7.613 | 10.943 | 5.295 | 138.41 | 292.814 |
| YNbO4:Eu3+ | 7.617 | 10.949 | 5.299 | 138.43 | 293.235 |
| YNbO4:Tb3+ | 7.615 | 10.946 | 5.298 | 138.42 | 293.03 |
| YNbO4:Eu3+,Tb3+ | 7.617 | 10.945 | 5.297 | 138.42 | 293.13 |
En el caso del niobato de itrio, la sustitución de átomos de itrio (Y) por iones de Eu3+ incrementó el volumen de la celda unitaria en 0.143%, en 0.074% para el reemplazo por iones de Tb3+ y en 0.108% cuando se sustituyeron simultáneamente por iones de Eu3+ y Tb3+.
Según el libro de Shannon 27, el radio iónico de los iones Eu3+ y Tb3+ es mayor que el radio iónico del Y. Debido a esto, el volumen de las celdas unitarias aumenta cuando se realiza dicha sustitución.
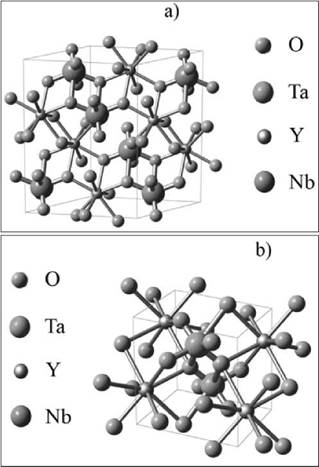
Para recrear las estructuras monoclínicas M'-YTaO4 y M-YNbO4 se utilizó el software libre Balls & Sticks 28 y los datos cristalográficos obtenidos al refinar dichas estructuras por medio de GSAS [29-30]. Estas imágenes se muestran en la figura 4.
En la figura 4a, los átomos de Y también están rodeados por 8 átomos de oxígeno (O) formando un cubo distorsionado. La distancia promedio encontrada entre los átomos de Y-O es de 2.35 Å. Los átomos de niobio (Nb) están en coordinación octaédrica distorsionada con cuatro enlaces cortos Nb-O, dos de 1.94 Å y dos de 1.83 Å y dos enlaces largos de 2.43 Å. En esta celda hay 4 átomos de itrio, 4 de niobio y 16 de oxígeno.
En la figura 4b, los átomos de Y están rodeados por 8 átomos de oxígeno formando una figura cúbica distorsionada. La distancia promedio entre los átomos de Y-O es 2.37 Å. Los átomos de Ta forman una figura octaédrica distorsionada con cuatro enlaces cortos de Ta-O, dos de 1.96 Å y dos de 1.87 Å, y dos enlaces largos de 2.23 Å. En esta celda hay 2 átomos de itrio, 2 de tantalio y 8 de oxígeno.
5. CONCLUSIONES
La caracterización morfológica de las estruc turas sintetizadas muestra que el tamaño pro medio de cristalitos obtenidos es ideal para la fabricación de pantallas intensificadoras de rayos X y pantallas de emisión de campo.
La introducción de defectos sustitucionales (átomos de Y por iones de Eu3+ y/o Tb3+) no alteró las características estructurales de los fósforos estudiados. Debido a la diferencia en los radios iónicos de los activadores Eu3+ y Tb3+ el volumen de la celda unitaria aumentó según la ley de Vegard.
Los datos cristalográficos de los fósforos M'- YTaO4 y M-YNbO4 se obtuvieron mediante el método de refinamiento Rietveld y están en concordancia con la literatura. Posteriormente se calculó los datos cristalográficos de las es tructuras dopadas con iones Eu3+ y Tb3+ y se recreó la estructura por medio del software libre Balls&Sticks.

