










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkNova
Print version ISSN 1794-2470
Nova vol.12 no.21 Bogotá Jan./June 2014
Trisomía 22 en un recién nacido de 39 semanas
Trisomy 22 in a newborn infant of 39 weeks
Ana María ávila1, Isabel Fernández2, Boris Linares1, María Luisa Quevedo1, Carlos David Aguana1, Luis Gustavo Celis1.
1. Facultad de Medicina-Universidad de La Sabana, Bogotá, Colombia.
2. Unidad de Genética Médica. Policlínica Metropolitana, Caracas, Venezuela.
Correo electrónico: luis.celis@unisabana.edu.co
Recibido: 08/04/2014Â Aceptado: 17/05/2014
RESUMEN
En este reporte presentamos el caso de un paciente masculino nacido de 39 semanas, producto de tercera gestación (dos abortos anteriores) de madre de 38 años y padre de 46 años. Las características clínicas del paciente incluyen macrocefalia, fontanela anterior amplia con diástasis de sutura sagital, escleras grisáceas, pabellones auriculares displásicos de implantación baja, raíz nasal corta, pliegue simiano en mano derecha e hirsutismo. Se obtienen tomografía axial computarizada de cráneo y resonancia magnética cerebral que presentan agenesia de cuerpo calloso y dilatación del asta occipital de los ventrículos laterales.
El cariotipo en sangre periférica evidencia trisomía parcial del cromosoma 22 (47, XY+22, del (22) (q11.2qter)). El paciente requirió 7 días de hospitalización y se da egreso hospitalario en buenas condiciones generales pero con un retardo psicomotor severo e hipotonía generalizada. Dadas las malformaciones estructurales severas que se presentan en este síndrome, los embarazos a término y la supervivencia postnatal de los niños con trisomía 22 son eventos muy raros. El caso de este paciente complementa otros reportes ilustrando que la trisomía 22 puede sobrevivir más allá del nacimiento.
Palabras clave: dimorfismo, malformaciones, resonancia magnética cerebral, tomografía axial, trisomía 22.
ABSTRACT
In this report, we present the case of a male patient who was born 39 weeks, the product of third gestation (two previous abortions) with a 38 year old mother and a 46 year old father. The clinical characteristics of the patient include macrocephaly, extensive anterior fontanelle with diastasis recti sagittal suture, ochronosis grayish pavilions dysplastic headphones lowset, short nasal root, simian crease in her right hand and hirsutism. We obtained a computerized axial tomography of skull and a brain magnetic resonance with agenesis of the corpus callosum and dilation of the ASTA occipital of the lateral ventricles.
The karyotype in peripheral blood evidence partial trisomy of chromosome 22 (47, XY+22, del (22) (q11.2qter)). The patient required 7 days of hospitalization and was released from the hospital in good condition overall, but with a psychomotor retardation and severe generalized hypotonia. Given the severe structural malformations that are present in this syndrome, the term pregnancy and post birth survival of children with trisomy 22 are very rare events. The case of this patient complements other reports illustrating that trisomy 22 can survive beyond birth.
Key Words: cerebral MRI, CT scan, dimorphism, malformations, trisomy 22
INTRODUCCIÓN
La Trisomía 22 es la tercera trisomía causante de abortos espontáneos en el primer trimestre de embarazo, sin embargo es un síndrome raro en nacidos vivos con una incidencia de 1/ 30.000-50.000 (1,2). El diagnóstico en el segundo o tercer trimestre es inusual, resulta del estudio de una restricción del crecimiento intrauterino con o sin malformaciones anatómicas y se caracteriza por retardo mental severo, dismorfismo facial y retraso en el neurodesarrollo con una esperanza de vida limitada (3).
De acuerdo a la literatura, el primer reporte de Trisomía 22 en un nacido vivo data de 1971 y se caracterizó por retardo mental severo, dismorfismo facial y retraso en el neurodesarrollo con una esperanza de vida limitada (4,5). En este artículo reportamos un caso de Trisomía 22 en un recién nacido de 39 semanas. Dadas las malformaciones estructurales severas que se presentan en este síndrome, los embarazos a término y la supervivencia postnatal de los niños con trisomía 22 son eventos muy raros. El caso de este paciente complementa otros reportes ilustrando que la Trisomía 22 puede sobrevivir más allá del nacimiento.
METODOLOGÍA
La metodología incluyó, además del examen físico, una entrevista a los padres, con el fin de elaborar la genealogía. También se realizó un cariotipo de linfocitos en sangre periférica con bandeo G. De la misma forma, se hizo una Tomografía Axial Computarizada (TAC) de cráneo y Resonancia Magnética Nuclear (RMN) cerebral, al igual que un Ecocardiograma Fetal.
RESULTADOS
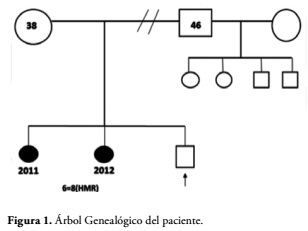
Se trata de un recién nacido masculino producto de tercera semana de gestación (IIIG). Madre de 38 años con dos abortos anteriores (IIA) y padre de 46 años, Figura 1. Los padres eran saludables, sin consanguineidad y sin antecedentes de enfermedad genética hereditaria hasta tres generaciones. Embarazo a término controlado, complicado con amenaza de parto pre término en el segundo trimestre, ameritando reposo absoluto por 3 semanas. A las 26 semanas se realizó ecocardiograma fetal que evidenció arritmia cardíaca.
Obtenido por cesárea segmentaria a las 39 semanas de gestación, lloró y respiró espontáneamente al nacer. Su peso fue de 2920 gramos, considerado adecuado para la edad (-1 DE) talla de 50 cm (0 DE), perímetro cefálico de 37,5 cm (> 2DE), perímetro torácico de 32 cm y perímetro abdominal de 30 cm.
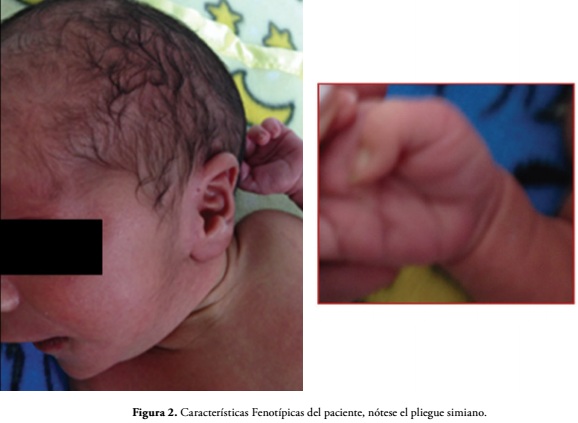
En el examen físico inicial se evidencia macrocefalia, asimetría craneofacial con cabalgamiento óseo, fontanela anterior amplia con diastasis de sutura sagital, hemangioma en línea media, escleras grisáceas, pabellones auriculares displásicos de implantación baja rotados, pits preauricular bilateral, raíz nasal corta con narinas ante vertidas, philtrum largo, microretrognatia, pliegue simiano en mano derecha, surcos plantares profundos e hirsutismo, Figura 2.
Con respecto al examen neurológico, se anota: succión débil, activo reactivo a estímulos exteroceptivos, reflejo rojo presente, hipotonía muscular, reflejos osteotendinosos presentes y simétricos, moro, prensión palmar y plantar presentes.
El recién nacido ingresa a la unidad neonatal para monitoreo de la arritmia cardíaca diagnosticada in útero además de presentar síndrome de dificultadrespiratoria (taquipnea transitoria del recién nacido). El paciente recibió manejo con oxígeno por cámara cefálica durante 3 días. Adicionalmente, presentó ictericia neonatal multifactorial ameritando fototerapia por 3 días.
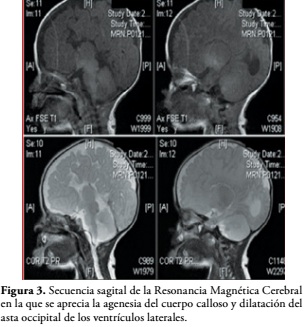
Durante su permanencia en la unidad neonatal se realizaron los siguientes estudios de extensión y de imágenes diagnósticas tales como TAC de cráneo simple y RMN cerebral que demuestran agenesia de cuerpo calloso y dilatación de las astas occipitales de los ventrículos laterales. La ecografía cerebral y abdominal fueron normales y el perfil tiroideo fue normal, Figura 3.
La evaluación cardiovascular evidenció bloqueo auriculoventricular de segundo grado tipo Mobitz II con frecuencia 2:1 con resolución espontánea. Ductus arterioso restrictivo y foramen oval permeable, que posteriormente desapareció.
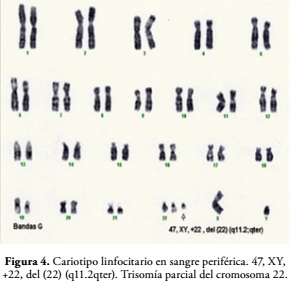
El Cariotipo linfocitario en sangre periférica mediante Técnica de Banda G mostró una composición cromosómica 47, XY, +22, del (22) (q11.2; qter). Trisomía parcial del cromosoma 22, Figura 4. El paciente requirió 7 días de hospitalización y se da su egreso en buenas condiciones generales, pero con un retardo psicomotor severo e hipotonía generalizada.
DISCUSIÓN
Al analizar la incidencia y el espectro de cromosomopatías en 2180 abortos espontáneos, se confirmó que la trisomía 16 era la más común causa de abortos espontáneos, seguida de cerca por la trisomía 21 y la trisomía 22, que representa el 11-16% de los casos (1). Descrita por primera vez en 1971 por Hsu, se estima que la trisomía 22 se presenta en 1/200 embarazos clínicamente reconocidos, lo que representa menos del 2,7% de abortos espontáneos y 0,2% de las muertes fetales (6,7).
El cromosoma 22 adicional se correlaciona con la edad materna y es el resultado (en un 96% de los casos) de la no disyunción materna durante la ovogénesis, en especial durante la primera división meiótica (8). Por otra parte, el 1,8% de los casos corresponden a alteraciones paternas o mitóticas, sin embargo no se ha comprobado que la supervivencia dependa del origen (9).
La no disyunción materna es causada por un proceso de recombinación alterado, se estima que el 25% del total de meiosis maternas I que derivan en trisomía 22 involucran cromosomas aquiasmáticos (8). Estos resultados son similares en la trisomía 21, los cromosomas 21 y 22 son los autosomas más pequeños y en promedio cada par está unido por uno o dos quiasmas durante la meiosis materna. Por consiguiente, la pérdida de un solo quiasma produce cromosomas aquiasmáticos (10).
A partir de 1980, gracias a la implementación de nuevas técnicas citogenéticas (entre ellas FISH), se han podido diferenciar trisomías 22 completas, parciales y translocaciones 11/22 (11). Estas últimas antes presumidas y catalogadas como trisomías 22, al igual que nos han permito detectar mosaicismos y disomías uniparentales (4).
Se ha demostrado que la trisomía 22 completa puede llegar a ser compatible con edades gestacionales tardías y supervivencias posteriores al nacimiento
(1). En los últimos 20 años se han reportado 30 casos de trisomía 22 no mosaico completa, de estos casos, 22 nacieron vivos con una esperanza de vida de 4 días (6). El periodo de supervivencia oscila entre minutos a 3 años de edad (12). La principales causas de muerte fueron falla cardiorrespiratoria o infecciones (13).
El diagnóstico prenatal de la trisomía 22 es infrecuente y existen pocos reportes al respecto (14). Los diagnósticos prenatales ecográficos son limitados, anomalías de las extremidades (reducción bilateral del fémur) y restricción del crecimiento intrauterino (RCIU) son los hallazgos más comúnmente encontrados en ecografías de tercer y segundo trimestre, en algunos casos hasta de primer trimestre (semanas 12 a 14) (15). Le siguen las alteraciones cardiacas presentes en un 86,7%, malformaciones cerebrales, anomalías renales y alteraciones craneofaciales, entre ellas, las auriculares se asocian comúnmente a cromosomopatías por lo que la evaluación de las orejas mediante ecografía o resonancia magnética fetal pueden ser de gran ayuda (15,16). La diferenciación entre trisomía 22 tipo mosaico y tipo no mosaico es importante principalmente con respecto a la esperanza de vida y posibles complicaciones siendo menores en casos tipo mosaico (17).
El caso que se describe es de trisomía 22 parcial, sin embargo el fenotipo puede llegar a ser similar al de la trisomía 22 completa, el cual se ha definido claramente (13). Entre sus características principales se destaca la hipoplasia facial con puente nasal plano y ancho, orejas displásicas con pits preauriculares, paladar hendido, hipertelorismo, microcefalia o alteraciones craneales, enfermedad cardiaca congénita, alteraciones urogenitales y RCIU (12,18). Por otra parte, en pacientes con trisomía 22 tipo mosaico se presenta frecuentemente hemiatrofia, defectos por reducción transversa de extremidades, sindactilia, hipomelanosis de Ito, hipoacusia neurosensorial o conductiva y retardo mental (12, 19, 20).
Adicionalmente, se han descrito casos de trisomía 22 tipo mosaico asociado a enfermedad de Hirschprung, alteraciones anatómicas a nivel del oído interno y tres casos de pacientes con desarrollo cognitivo normal (18, 21-23). Por consiguiente, es posible que existan casos de trisomía 22 tipo mosaico con mínimos hallazgos al examen físico y desarrollo cognitivo adecuado que no sean diagnosticados (24).
Otro aspecto importante es que las trisomías 13, 8 y 22 presentan algunas características dismórficas comunes tales como anormalidades en el cráneo, orejas displásicas, nariz corta, paladar hendido, cuello corto y uñas hipoplásicas (25), por lo que es muy importante los análisis citogenéticos para confirmar el diagnóstico de la enfermedad como el caso estudiado.
Dadas las malformaciones estructurales severas que se presentan en este síndrome, los embarazos a término y la supervivencia postnatal de los niños con trisomía 22 son eventos muy raros. No obstante, un mosaicismo no detectado confinado a la placenta o en tejidos fetales diferentes a sangre o piel podría explicar los pocos casos de supervivencia que se han reportado (1). El presente caso complementa otros reportes ilustrando que la trisomía 22 parcial puede sobrevivir más allá del nacimiento. A pesar de los dilemas éticos, en cuanto a la realización de un tamizaje con pruebas diagnósticas prenatales complementarias, es imperativa la realización de asesoramiento genético a la familia.
Actualmente se realizan estudios citogenéticos en vellosidades coriónicas y líquido amniótico en pacientes con alto riesgo (edad materna avanzada, hallazgos anormales en la ecografía, alteraciones en paraclínicos séricos o historia personal y/o familiar de cromosomopatía) (12, 26). Por último, es necesario resaltar que estos pacientes necesitan el apoyo de un equipo multidisciplinario para lograr conseguir la mejor calidad de vida para ellos y su grupo familiar, en el caso estudiado el paciente continúa con vida pero con retardo psicomotor severo e hipotonía generalizada.
Finalmente, es importante anotar que el permiso para el uso de la información utilizada en este reporte de caso ha sido dado por la Unidad de Genética Médica donde el paciente ha sido tratado. De la misma forma, se contó con el respectivo consentimiento informado.
REFERENCIAS
1. Heinrich T, Nanda I, Rehn M, et al. Liveborn trisomy 22: patient report and review. Mol Syndromol. 2013; 3(6):262-269. [ Links ]
2. Basaran N, Berkil H, Ay N, et al. A rare case: mosaic trisomy 22. Ann Genet. 2001; 44(4):183-186. [ Links ]
3. Mokate T, Leask K, Mehta S, et al. Nonmosaic trisomy 22: a report of 2 cases. Prenat Diagn. 2006; 26(10):962-965. [ Links ]
4. Crowe CA, Schwartz S, Black CJ, Jaswaney V. Mosaic trisomy 22: a case presentation and literature review of trisomy 22 phenotypes. Am J Med Genet. 1997; 71(4):406-413. [ Links ]
5. Vaglio A, Milunsky A, Huang XL, et al. A 21 years follow up of a girl patient with a pseudodicentric bisatellited chromosome 22 associated with partial trisomy 22pter->22q12.1: clinical, cytogenetic and molecular observations. Eur J Med Genet. 2008; 51(4):332-342. [ Links ]
6. Hassold TJ, Jacobs PA. Trisomy in man. Annu Rev Genet. 1984; 18:69-97. [ Links ]
7. Shih JC, Shyu MK, Lee CN, Wu CH, Lin GJ, Hsieh FJ. Antenatal depiction of the fetal ear with threedimensional ultra-sonography. Obstet Gynecol. 1998; 91(4):500-505. [ Links ]
8. Hall HE, Surti U, Hoffner L, Shirley S, Feingold E, Hassold T. The origin of trisomy 22: evidence for acrocentric chromosome-specific patterns of nondisjunction. Am J Med Genet A. 2007; 143A(19):2249-2255. [ Links ]
9. Mihçi E, Taçoy S, Yakut S, et al. Maternal origin and clinical findings in a case with trisomy 22. Turk J Pediatr. 2007; 49(3):322-326. [ Links ]
10. Lamb NE, Feingold E, Savage A, et al. Characterization of susceptible chiasma configurations that increase the risk for maternal nondisjunction of chromosome 21. Hum Mol Genet. 1997; 6(9):1391-1399. [ Links ]
11. Thomas S, Parker M, Tan J, Duckett D, Woodruff G. Ocular manifestations of mosaic trisomy 22: a case report and review of the literature. Ophthalmic Genet. 2004; 25(1):53-56. [ Links ]
12. Rao VB, Seema K, Lily K, Ghosh K, Mohanty D. Trisomy 22 with unusual phenotype. Indian Pediatr. 2003; 40(4): 371-372. [ Links ]
13. Tinkle BT, Walker ME, Bloughpfau RI, Saal HM, Hopkin RJ. Unexpected survival in a case of prenatally diagnosed non-mosaic trisomy 22: Clinical report and review of the natural history. Am J Med Genet A. 2003; 118 A(1):90-95. [ Links ]
14. Milic A, Blaser S, Robinson A, et al. Prenatal detection of microtia by MRI in a fetus with trisomy 22. Pediatr Radiol. 2006; 36(7):706-710. [ Links ]
15. Stressig R, Körtge-jung S, Hickmann G, Kozlowski P. Prenatal sonographic findings in trisomy 22: five case reports and review of the literature. J Ultrasound Med. 2005; 24(11):1547-1553. [ Links ]
16. Shih JC, Shyu MK, Lee CN, Wu CH, Lin GJ, Hsieh FJ. Antenatal depiction of the fetal ear with three-dimensional ultra-sonography. Obstet Gynecol. 1998; 91(4):500-505. [ Links ]
17. Leclercq S, Baron X, Jacquemont ML, Cuillier F, Cartault F. Mosaic trisomy 22: five new cases with variable outcomes. Implications for genetic counselling and clinical management. Prenat Diagn. 2010; 30(2):168-172. [ Links ]
18. Abdelgadir D, Nowaczyk MJ, Li C. Trisomy 22 mosaicism and normal developmental outcome: report of two patients and review of the literature. Am J Med Genet A. 2013;161 A (5):1126-1131. [ Links ]
19. Woods CG, Bankier A, Curry J, et al. Asymmetry and skin pigmentary anomalies in chromosome mosaicism. J Med Genet. 1994; 31(9):694-701. [ Links ]
20. Ruiter EM, Toorman J, Hochstenbach R, De Vries BB. Mosaic trisomy 22 in a boy with a terminal transverse limb reduction defect. Clin Dysmorphol. 2004; 13(2):99-102. [ Links ]
21. Hall T, Samuel M, Brain J. Mosaic trisomy 22 associated with total colonic aganglionosis and malrotation. J Pediatr Surg. 2009; 44(1):e9-e11. [ Links ]
22. Florez L, Lacassie Y. Mosaic trisomy 22: report of a patient with normal intelligence. Am J Med Genet A. 2005; 132A(2):223-225. [ Links ]
23. Ohtani I, Kano M, Sagawa Y, Ogawa H, Suzuki C. Temporal bone histopathology in trisomy 22. Int J Pediatr Otorhinolaryngol. 2001; 59(2):137-141. [ Links ]
24. Lewis B, Fulton S, Short E, et al. A longitudinal case study of a child with mosaic trisomy 22: language, cognitive, behavioral, physical, and dental outcomes. Am J Med Genet A. 2007; 143A(17):2070-2074. [ Links ]
25. Naicker T, Aldous C. Two Trisomy 22 Live Births in One Hospital in 15 Months: Is It as Rare as We Thought? Fetal and Pediatric Pathology 2014; 33:35-41. [ Links ]
26. Vora NL, O'brien BM. Noninvasive prenatal testing for microdeletion syndromes and expanded trisomies: proceed with caution. Obstet Gynecol. 2014; 123(5):1097-1099. [ Links ]
